przyczyny/dziedziczenie
co powoduje choroby mitochondrialne?
miopatie mitochondrialne są stosunkowo częste. Pierwotne zaburzenia mitochondrialne są najczęstszymi dziedzicznymi błędami metabolizmu. Częstość występowania encefalomiopatii mitochondrialnej u dzieci w wieku przedszkolnym wynosi 1 na 11 000. Choroba mitochondrialna spowodowana mutacjami w mitochondrialnym DNA ma szacunkową częstość występowania 1 na 5000. Jednak choroba mitochondrialna spowodowana mutacjami w DNA jądrowym ma szacunkową częstość występowania 1 na 35 000.,1
choroby mitochondrialne nie są zaraźliwe i nie są spowodowane przez nic, co człowiek robi. Są one spowodowane mutacjami lub zmianami w genach-planach komórek do wytwarzania białek.
geny są odpowiedzialne za budowę naszego ciała i są przekazywane z rodziców na dzieci, wraz z wszelkimi mutacjami lub defektami, które mają. Oznacza to, że choroby mitochondrialne są dziedziczne, chociaż często wpływają na członków tej samej rodziny w różny sposób. (Więcej informacji na temat mutacji genetycznych i chorób mitochondrialnych można znaleźć poniżej.,)
geny biorące udział w chorobach mitochondrialnych zwykle wytwarzają białka działające w mitochondriach lub na ich powierzchni. W obrębie każdego mitochondrium (pojedyncza forma mitochondriów) białka te stanowią część linii montażowej, która wykorzystuje cząsteczki paliwa pochodzące z żywności do produkcji cząsteczki energii adenozynotrójfosforanu (ATP) w procesie zwanym fosforylacją oksydacyjną. Ten wysoce wydajny proces produkcji wymaga tlenu; poza mitochondriami istnieją mniej wydajne sposoby wytwarzania ATP bez tlenu.,
białka na początku mitochondrialnej linii montażowej działają jak ładowarki, importując cząsteczki paliwa — cukry i tłuszcze — do mitochondriów. Następnie inne białka rozkładają cukry i tłuszcze, wydobywając energię w postaci naładowanych cząstek zwanych elektronami.
białka na końcu linii — zorganizowane w pięć grup zwanych kompleksami I, II, III, IV I V oraz dwa ruchome nośniki elektronów, koenzym Q10 i cytochrom c — wykorzystują energię z tych elektronów do wytwarzania ATP., Kompleksy od I do IV przenoszą elektrony w dół linii i dlatego nazywane są łańcuchem transportu elektronów, a kompleks V faktycznie wypiera ATP, więc nazywany jest również syntazą ATP.
niedobór jednego lub więcej z tych kompleksów jest typową przyczyną choroby mitochondrialnej. (W rzeczywistości choroby mitochondrialne są czasami nazywane specyficznym niedoborem, takim jak niedobór kompleksu I.) Niedobór koenzymu Q10 spowodowany mutacjami DNA jądrowego może objawiać się osłabieniem mięśni proksymalnych w izolacji., Aby prawidłowo funkcjonować, białka szlaku fosforylacji oksydacyjnej muszą zostać przetłumaczone, zaimportowane do mitochondriów i wprowadzone do wewnętrznej błony mitochondrialnej. Mutacje w genach wpływających na te procesy mogą również powodować miopatię mitochondrialną.
wady genów, które są związane ze strukturą i dynamiką mitochondriów, mogą być również zaangażowane w rozwój miopatii., Kiedy komórka jest wypełniona wadliwymi mitochondriami, nie tylko zostaje pozbawiona ATP, ale także może gromadzić zaległości niewykorzystanych cząsteczek paliwa i tlenu, co może mieć katastrofalne skutki.
w takich przypadkach nadmiar cząsteczek paliwa jest wykorzystywany do wytwarzania ATP za pomocą nieefektywnych środków, które mogą generować potencjalnie szkodliwe produkty uboczne, takie jak kwas mlekowy. (Dzieje się tak również wtedy, gdy komórka ma niewystarczający dopływ tlenu, co może się zdarzyć do komórek mięśniowych podczas forsownych ćwiczeń.,) Nagromadzenie kwasu mlekowego we krwi-zwane kwasicą mleczanową – wiąże się ze zmęczeniem mięśni i może faktycznie uszkodzić tkankę mięśniową i nerwową.
w międzyczasie niewykorzystany tlen w komórce może zostać przekształcony w destrukcyjne związki zwane reaktywnymi formami tlenu, w tym tak zwanymi wolnymi rodnikami. (Są to cele leków przeciwutleniających i witamin.)
ATP pochodzący z mitochondriów stanowi główne źródło energii do skurczu komórek mięśniowych i wypalania komórek nerwowych. Tak więc komórki mięśniowe i komórki nerwowe są szczególnie wrażliwe na wady mitochondrialne., Połączone skutki deprywacji energii i akumulacji toksyn w tych komórkach prawdopodobnie powodują główne objawy miopatii mitochondrialnej i encefalomiopatii.
podsumowując, w zależności od pierwotnej wady genetycznej choroby mitochondrialne mogą być spowodowane przez zmiany: białek łańcucha oddechowego, białek pomocniczych łańcucha oddechowego, translacji mitochondrialnego RNA, środowiska lipidowego błony wewnętrznej mitochondrialnej, zubożenia mitochondrialnego DNA i dynamiki mitochondrialnej., Miopatie mitochondrialne można również podzielić na kategorie w zależności od tego, który z tych procesów jest zmieniony.
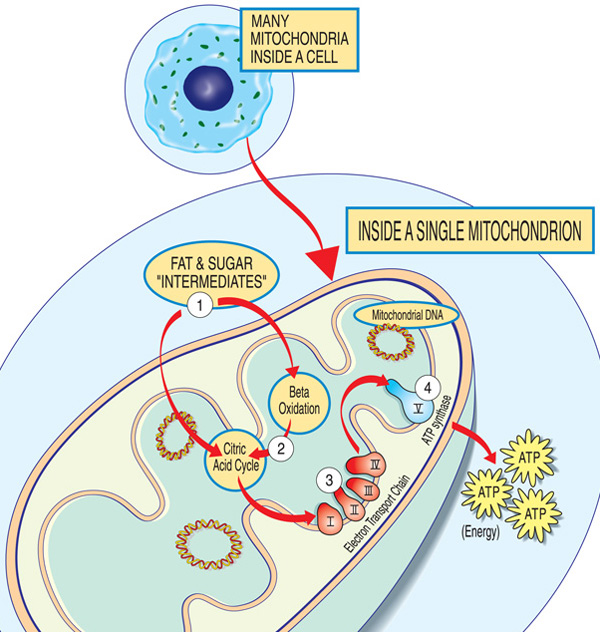
każdy mitochondrion jest fabryką energii, która „importuje” cukry i tłuszcze, rozkłada je i „eksportuje” energię (ATP) poprzez następujące czynności: tłuszcze i półprodukty cukrowe dostają się do mitochondriów. Kwasy tłuszczowe są rozkładane przez utlenianie beta i usuwanie elektronów w cyklu kwasu cytrynowego. Elektrony są przepuszczane przez główny kompleks łańcucha transportu elektronów. ATP jest wytwarzany przez syntazę ATP.,
jakie są wzorce dziedziczenia w miopatiach mitochondrialnych?
genetyka mitochondrialna jest złożona i często choroba mitochondrialna może być trudna do prześledzenia przez drzewo genealogiczne. Ale ponieważ są one spowodowane przez wadliwe geny, choroby mitochondrialne nie uruchomić w rodzinach.
aby zrozumieć, w jaki sposób choroby mitochondrialne są przenoszone przez rodziny, ważne jest, aby wiedzieć, że istnieją dwa rodzaje genów niezbędnych dla mitochondriów. Pierwszy typ jest umieszczony w jądrze-część naszych komórek, która zawiera większość naszego materiału genetycznego lub DNA., Drugi typ zamieszkuje wyłącznie DNA zawarte wewnątrz samych mitochondriów.
mutacje DNA jądrowego (nDNA) lub DNA mitochondrialnego (mtDNA) mogą powodować choroby mitochondrialne.
Większość nDNA (wraz z jakąkolwiek mutacją) jest dziedziczona we wzorze Mendla, co luźno oznacza, że jedna kopia każdego genu pochodzi od każdego rodzica. Ponadto większość chorób mitochondrialnych wywołanych przez mutacje nDNA (w tym zespół Leigh, MNGIE, a nawet MDS) jest autosomalnie recesywna, co oznacza, że do wywołania choroby potrzeba mutacji w obu kopiach genu.,
w przeciwieństwie do nDNA, mtDNA przechodzi tylko z matki na dziecko. To dlatego, że podczas zapłodnienia, kiedy plemniki łączą się z jajkiem, mitochondria plemników-i jego mtDNA – są niszczone. Tak więc choroby mitochondrialne wywołane mutacjami mtDNA są unikalne, ponieważ są dziedziczone we wzorze matczynym(patrz ilustracja poniżej).
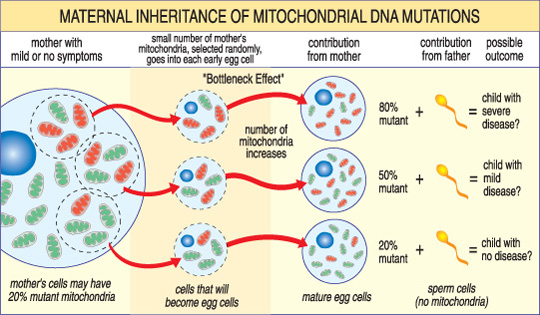
nasilenie choroby mitochondrialnej u dziecka zależy od odsetka nieprawidłowych (zmutowanych) mitochondriów w komórce jajowej, która je utworzyła.,
kolejna unikalna cecha chorób mtDNA wynika z faktu, że typowa komórka ludzka — w tym komórka jajowa — zawiera tylko jedno jądro, ale setki mitochondriów. Pojedyncza komórka może zawierać zarówno zmutowane mitochondria, jak i normalne mitochondria, a równowaga między nimi determinuje zdrowie komórki. Pomaga to wyjaśnić, dlaczego objawy choroby mitochondrialnej mogą się tak bardzo różnić w zależności od osoby, nawet w obrębie tej samej rodziny.,
wyobraź sobie, że komórki jajowe kobiety (i inne komórki w jej ciele) zawierają zarówno normalne, jak i zmutowane mitochondria, a niektóre mają tylko kilka zmutowanych mitochondriów, podczas gdy inne mają wiele. Dziecko poczęte z” głównie zdrowej ” komórki jajowej prawdopodobnie nie rozwinie choroby, a dziecko poczęte z „głównie zmutowanej” komórki jajowej prawdopodobnie tak będzie. Ponadto, kobieta może lub nie może mieć objawy choroby mitochondrialnej siebie.
choroby te również mogą pojawić się sporadycznie, co oznacza, że mogą wystąpić bez historii rodziny.,
ryzyko przeniesienia choroby mitochondrialnej na dzieci zależy od wielu czynników, w tym od tego, czy choroba jest spowodowana mutacjami w nDNA czy mtDNA. Dobrym sposobem, aby dowiedzieć się więcej o tych zagrożeniach, jest rozmowa z lekarzem lub doradcą genetycznym w Centrum Opieki MDA. Zobacz również fakty na temat genetyki i chorób nerwowo-mięśniowych.
Leave a Reply