Årsakene/Arv
Hva er årsaken til mitokondrie sykdommer?
Mitokondrie myopathies er relativt vanlig. Primære mitokondrie lidelser er de vanligste arvelig feil i stoffskiftet. Utbredelsen av mitokondrie encephalomyopathies for førskole-alderen barn er 1 i 11,000. Mitokondrie sykdom forårsaket av mutasjoner i mitokondrie-DNA, har en estimert prevalens av 1 5000. Men mitokondrie sykdom forårsaket av mutasjoner i det kjernefysiske DNA har en estimert prevalens på 1 i 35,000.,1
Mitokondrie sykdommer er ikke smittsom, og de er ikke forårsaket av noe en person gjør. De er forårsaket av mutasjoner eller endringer i gener — cellene’ tegninger for å lage proteiner.
Gener er ansvarlig for å bygge kroppen vår, og er gått fra foreldre til barn, sammen med eventuelle mutasjoner eller mangler de har. Det betyr at mitokondrienes sykdommer er arvbare, selv om de ofte påvirke medlemmer av samme familie på forskjellige måter. (For mer informasjon om genetiske mutasjoner og mitokondrie sykdom, se nedenfor.,)
gener involvert i mitokondrie sykdom gjør vanligvis proteiner som arbeider i eller på mitokondriene. Innenfor hver mitokondrium (form entall av mitokondrier), disse proteinene utgjør en del av et samlebånd som bruker drivstoff molekyler som er avledet fra mat til å produsere energi molekylet adenosintrifosfat (ATP) gjennom en prosess som heter oksidativt phosphorylation. Denne svært effektive produksjonsprosessen krever oksygen; utenfor mitokondrium, det er mindre effektive måter å produsere ATP uten oksygen.,
Proteiner i begynnelsen av mitokondrie samlebåndet opptre som last handlere, importere drivstoff molekyler — sukker og fett — i mitokondrie. Neste, andre proteiner bryte ned sukker og fett, trekke ut energi i form av ladede partikler som kalles elektroner.
Proteiner mot slutten av linjen — organisert i fem grupper kalt komplekser i, II, III, IV og V, og to mobile electron bærere, koenzym Q10 og cytokrom c — utnytte energien fra disse elektronene til å lage ATP., Komplekser jeg gjennom IV transport av elektroner ned linjen, og er derfor kalt elektrontransportkjeden, og komplekse V faktisk spyr ut ATP, så det er også kalt ATP syntase.
En mangel på ett eller flere av disse kompleksene er den vanligste årsaken til en mitokondrie sykdom. (Faktisk, mitokondrie sykdommer er noen ganger kalt for en spesifikk mangel, for eksempel komplekse jeg mangel.) Koenzym Q10-mangel på grunn av kjernefysiske DNA-mutasjoner kan presentere med proksimal muskelsvakhet i isolasjon., For å fungere på riktig måte, proteiner av oksidativt phosphorylation veien må være oversatt, importert inn i mitokondriene, og satt inn i mitokondrienes indre membran. Mutasjoner i gener som påvirker disse prosessene kan også føre til mitokondrie myopati.
Defekter i gener som er knyttet til strukturen og dynamikken i mitokondriene kan også være involvert i utviklingen av myopathies., Når en celle er fylt med defekt mitokondrier, ikke bare det å bli fratatt ATP, det kan også samle seg opp et etterslep av ubrukte drivstoff molekyler og oksygen, med potensielt katastrofale virkninger.
I slike tilfeller overflødig drivstoff molekyler som er brukt til å lage ATP ved ineffektiv måte, noe som kan gi potensielt skadelige biprodukter som melkesyre. (Dette skjer også når en celle har en utilstrekkelig oksygentilførsel, noe som kan skje til muskelcellene under anstrengende trening.,) Opphoping av melkesyre i blodet — som kalles melkesyreacidose — er forbundet med muskel tretthet og kan faktisk skade muskel-og nervevev.
i Mellomtiden, ubrukt oksygen i cellen kan konverteres til ødeleggende stoffer som kalles reaktive oksygen arter, inkludert såkalte frie radikaler. (Disse målene av antioksidant medisiner og vitaminer.)
ATP avledet fra mitokondrier gir den viktigste kilden til makt for muskel celle sammentrekning og nervecelle avfyring. Så, muskelceller og nerveceller er spesielt følsomme for mitokondrie feil., Den kombinerte effekten av energi deprivasjon og toksin akkumulering i disse cellene trolig gi opphav til de viktigste symptomene på mitokondrie myopathies og encephalomyopathies.
I sammendrag, avhengig av den primære genetiske defekter, mitokondrie sykdommer som kan være forårsaket av endringer av følgende: luftveier kjede proteiner, luftveier kjede hjelpeutstyr proteiner, mitokondrie-RNA oversettelse, mitokondrienes indre membran lipid miljø, reduksjon av mitokondrie-DNA, og mitokondrie dynamics., Mitokondrie myopathies kan også være delt inn i kategorier basert på hvilke av disse prosessene er endret.
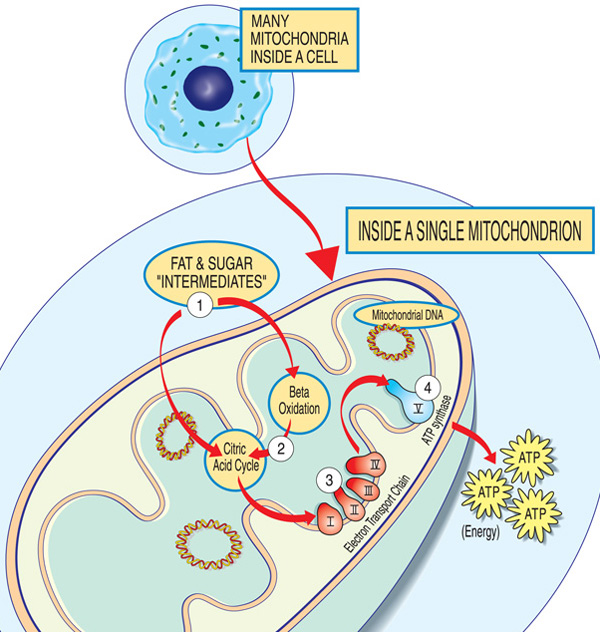
Hver mitokondrium er en energi-fabrikken som «import» sukker og fett, bryter dem ned og «eksport» energi (ATP) via disse trinnene: Fett og sukker intermediater angi mitokondrium. Fettsyrer er brutt ned gjennom beta-oksidasjon og fjerning av elektroner i sitronsyre syklus. Elektroner er gått gjennom store kompleks av elektrontransportkjeden. ATP er laget av ATP syntase.,
Hva er arv mønstre i mitokondrie myopathies?
Mitokondrie genetikk er komplekse, og ofte, en mitokondrie sykdommen kan være vanskelig å spore gjennom et familietre. Men fordi de er forårsaket av defekte gener, mitokondrie sykdommer må kjøre i familier.
for Å forstå hvordan mitokondrie sykdommer er overlevert gjennom familier, er det viktig å vite at det finnes to typer av gener som er avgjørende for å mitokondrier. Den første typen er plassert i kjernen — delen av våre celler som inneholder det meste av vårt genetiske materiale, eller DNA., Den andre typen er bosatt utelukkende innenfor DNA som finnes inne i mitokondriene seg selv.
Mutasjoner i enten kjernefysiske DNA (nDNA) eller mitokondrie DNA (mtDNA) kan føre til mitokondrie sykdom.
de Fleste nDNA (sammen med eventuelle mutasjoner det har) er arvelig i en Mendelian mønster, løst noe som betyr at en kopi av hvert gen kommer fra hver av foreldrene. Også de fleste mitokondrie sykdommer forårsaket av nDNA mutasjoner (inkludert Leigh syndrom, MNGIE, og selv MDS) er autosomal recessiv, noe som betyr at det tar mutasjoner i begge kopier av et gen til å forårsake sykdom.,
i Motsetning til nDNA, mtDNA går bare fra mor til barn. Det er fordi under unnfangelsen, når sperm sikringer med egg, sperm er mitokondrier — og sitt mtDNA — er ødelagt. Dermed mitokondrie sykdommer forårsaket av mtDNA mutasjoner er unike fordi de er arvet i en mors mønster (se illustrasjon nedenfor).
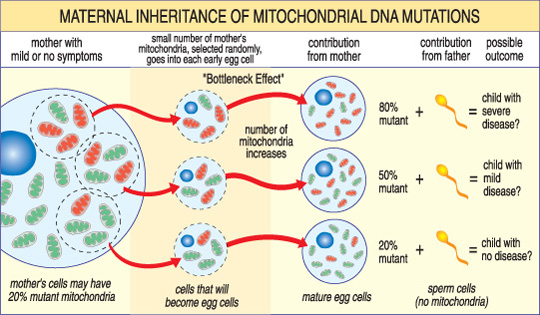
alvorlighetsgraden av en mitokondrie sykdom hos et barn avhenger av hvor stor prosentandel av unormal (mutant) mitokondrier i egg celle som dannet dem.,
en Annen unik funksjon i mtDNA sykdommer oppstår fra det faktum at en typisk menneskelig celle — inkludert egg celle — inneholder bare én kjerne, men hundrevis av mitokondrier. En enkelt celle kan inneholde både mutant mitokondrier og normal mitokondrier, og balansen mellom de to avgjør cellens helse. Dette bidrar til å forklare hvorfor symptomene på mitokondrie sykdom kan variere så mye fra person til person, selv innen samme familie.,
Tenk deg at en kvinnes egg celler (og andre celler i kroppen hennes) inneholder både normal og mutant mitokondrier, og at noen har bare et par mutant mitokondrier, mens andre har mange. Et barn unnfanget fra et «stort sett sunn» egg celle sannsynligvis vil ikke utvikle sykdommen, og et barn unnfanget fra et «stort sett mutant» egg celle trolig vil. Også, kvinnen kan eller ikke kan ha symptomer på mitokondrie sykdommen selv.
Disse sykdommene kan også oppstå i en sporadiske mote, noe som betyr at de kan oppstå med ingen familie historie.,
risikoen for å formidle en mitokondrie sykdom til barn avhenger av mange faktorer, blant annet om sykdommen er forårsaket av mutasjoner i nDNA eller mtDNA. En god måte å finne ut mer om disse farene er å snakke med en lege eller genetisk rådgiver på en MDA Care Center. Se også Fakta Om Genetikk og Nevromuskulære Sykdommer.
Leave a Reply