Causes/Hérédité
quelles sont les causes des maladies mitochondriales?
les myopathies Mitochondriales sont relativement fréquentes. Les troubles mitochondriaux primaires sont les erreurs héréditaires les plus courantes du métabolisme. La prévalence des encéphalomyopathies mitochondriales chez les enfants d’âge préscolaire est de 1 sur 11 000. La maladie mitochondriale causée par des mutations dans l’ADN mitochondrial a une prévalence estimée à 1 sur 5 000. Cependant, la maladie mitochondriale causée par des mutations dans l’ADN nucléaire a une prévalence estimée à 1 sur 35 000.,1
Les maladies mitochondriales ne sont pas contagieuses et ne sont pas causées par tout ce qu’une personne fait. Ils sont causés par des mutations ou des changements dans les gènes — cellules plans pour faire des protéines.
Les gènes sont responsables de la construction de notre corps et sont transmis des parents aux enfants, avec toutes les mutations ou défauts qu’ils ont. Cela signifie que les maladies mitochondriales sont héritables, bien qu’elles affectent souvent les membres de la même famille de différentes manières. (Pour plus d’informations sur les mutations génétiques et les maladies mitochondriales, voir ci-dessous.,)
Les gènes impliqués dans la maladie mitochondriale produisent normalement des protéines qui agissent dans ou sur les mitochondries. Au sein de chaque mitochondrie (la forme singulière des mitochondries), ces protéines font partie d’une chaîne d’assemblage qui utilise des molécules de carburant dérivées des aliments pour fabriquer la molécule d’énergie adénosine triphosphate (ATP) par un processus nommé phosphorylation oxydative. Ce processus de fabrication très efficace nécessite de l’oxygène; en dehors des mitochondries, il existe des moyens moins efficaces de produire de l’ATP sans oxygène.,
Les protéines au début de la chaîne d’assemblage mitochondriale agissent comme des manipulateurs de cargaison, important les molécules de carburant — sucres et graisses — dans la mitochondrie. Ensuite, d’autres protéines décomposent les sucres et les graisses, extrayant de l’énergie sous forme de particules chargées appelées électrons.
Les protéines vers la fin de la ligne — organisées en cinq groupes appelés complexes I, II, III, IV et V, et deux transporteurs d’électrons mobiles, la coenzyme Q10 et le cytochrome c — exploitent l’énergie de ces électrons pour produire de l’ATP., Les Complexes I à IV transfèrent les électrons le long de la ligne et sont donc appelés la chaîne de transport d’électrons, et le complexe V produit en fait de l’ATP, il est donc également appelé ATP synthase.
Une carence en un ou plusieurs de ces complexes est la cause typique d’une maladie mitochondriale. (En fait, les maladies mitochondriales sont parfois nommées pour une carence spécifique,telle que la carence en complexe I.) La carence en Coenzyme Q10 due à des mutations nucléaires de l’ADN peut se présenter avec une faiblesse musculaire proximale isolément., Pour fonctionner correctement, les protéines de la voie de phosphorylation oxydative doivent être traduites, importées dans les mitochondries et insérées dans la membrane mitochondriale interne. Les Mutations dans les gènes qui affectent ces processus peuvent également provoquer une myopathie mitochondriale.
Les défauts des gènes liés à la structure et à la dynamique des mitochondries peuvent également être impliqués dans le développement des myopathies., Lorsqu’une cellule est remplie de mitochondries défectueuses, non seulement elle est privée d’ATP, mais elle peut également accumuler un arriéré de molécules de carburant et d’oxygène inutilisés, avec des effets potentiellement désastreux.
dans de tels cas, des molécules de carburant en excès sont utilisées pour fabriquer de l’ATP par des moyens inefficaces, ce qui peut générer des sous-produits potentiellement nocifs tels que l’acide lactique. (Cela se produit également lorsqu’une cellule a un apport insuffisant en oxygène, ce qui peut arriver aux cellules musculaires pendant un exercice intense.,) L’accumulation d’acide lactique dans le sang — appelée acidose lactique-est associée à la fatigue musculaire et pourrait endommager les tissus musculaires et nerveux.
pendant ce temps, l’oxygène inutilisé dans la cellule peut être converti en composés destructeurs appelés espèces réactives de l’oxygène, y compris les radicaux libres. (Ce sont les cibles des médicaments antioxydants et des vitamines.)
L’ATP dérivé des mitochondries fournit la principale source d’énergie pour la contraction des cellules musculaires et la mise à feu des cellules nerveuses. Ainsi, les cellules musculaires et les cellules nerveuses sont particulièrement sensibles aux défauts mitochondriaux., Les effets combinés de la privation d’énergie et de l’accumulation de toxines dans ces cellules donnent probablement lieu aux principaux symptômes des myopathies mitochondriales et des encéphalomyopathies.
En résumé, selon le défaut génétique primaire, les maladies mitochondriales peuvent être causées par des altérations des éléments suivants: protéines de la chaîne respiratoire, protéines auxiliaires de la chaîne respiratoire, traduction de l’ARN mitochondrial, milieu lipidique de la membrane interne mitochondriale, épuisement de l’ADN mitochondrial et dynamique mitochondriale., Les myopathies mitochondriales peuvent également être divisées en catégories en fonction de laquelle de ces processus est altérée.
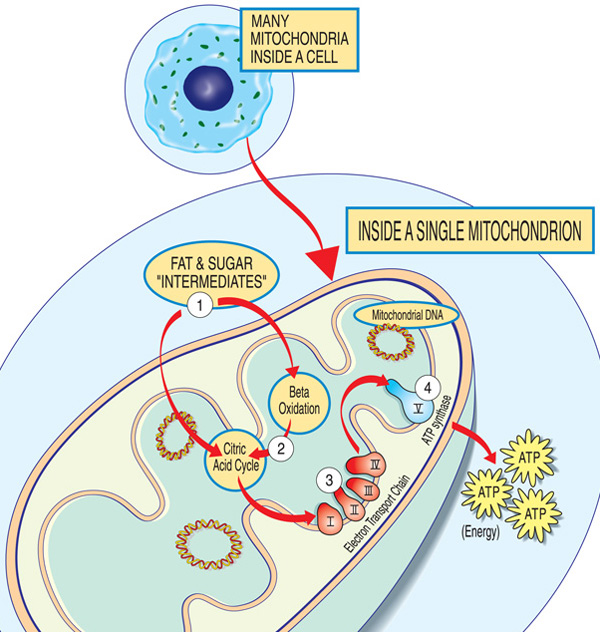
chaque mitochondrie est une usine énergétique qui « importe” des sucres et des graisses, les décompose et « exporte” de l’énergie (ATP) via ces étapes: les intermédiaires de graisse et de sucre entrent dans la mitochondrie. Les acides gras sont décomposés par oxydation bêta et l’élimination des électrons dans le cycle de l’acide citrique. Les électrons sont passés à travers le complexe majeur de la chaîne de transport d’électrons. L’ATP est fabriqué par L’ATP synthase.,
Quels sont les schémas d’hérédité dans les myopathies mitochondriales?
la génétique mitochondriale est complexe, et souvent, une maladie mitochondriale peut être difficile à tracer à travers un arbre généalogique. Mais parce qu’ils sont causés par des gènes défectueux, les maladies mitochondriales courent dans les familles.
pour comprendre comment les maladies mitochondriales sont transmises par les familles, il est important de savoir qu’il existe deux types de gènes essentiels aux mitochondries. Le premier type est logé dans le noyau – la partie de nos cellules qui contient la majeure partie de notre matériel génétique, ou ADN., Le deuxième type réside exclusivement dans L’ADN contenu à l’intérieur des mitochondries elles-mêmes.
des Mutations dans L’ADN nucléaire (adnn) ou l’ADN mitochondrial (ADNmt) peuvent causer des maladies mitochondriales.
la plupart des nDNA (ainsi que toutes les mutations qu’ils ont) sont hérités selon un schéma mendélien, ce qui signifie vaguement qu’une copie de chaque gène provient de chaque parent. En outre, la plupart des maladies mitochondriales causées par des mutations nDNA (y compris le syndrome de Leigh, MNGIE et même MDS) sont autosomiques récessives, ce qui signifie qu’il faut des mutations dans les deux copies d’un gène pour causer la maladie.,
contrairement à l’adnn, l’ADNmt ne passe que de la mère à l’enfant. En effet, lors de la conception, lorsque le sperme fusionne avec l’ovule, les mitochondries du sperme — et son ADNmt — sont détruites. Ainsi, les maladies mitochondriales causées par des mutations de l’ADNmt sont uniques parce qu’elles sont héritées selon un schéma maternel (voir illustration ci-dessous).
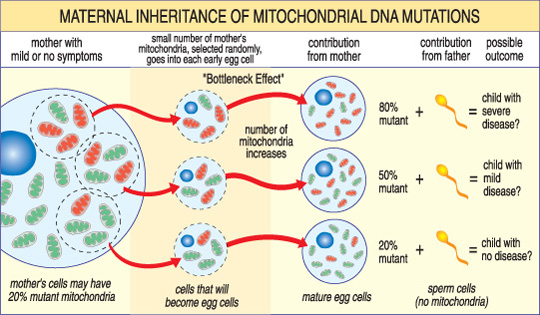
La gravité d’une maladie mitochondriale chez un enfant dépend du pourcentage de mitochondries anormales (mutantes) dans l’ovule qui les a formées.,
Une autre caractéristique unique des maladies de l’ADNmt provient du fait qu’une cellule humaine typique — y compris l’ovule — ne contient qu’un noyau mais des centaines de mitochondries. Une seule cellule peut contenir à la fois des mitochondries mutantes et des mitochondries normales, et l’équilibre entre les deux déterminera la santé de la cellule. Cela aide à expliquer pourquoi les symptômes de la maladie mitochondriale peuvent varier tant d’une personne à l’autre, même au sein d’une même famille.,
Imaginez que les ovocytes d’une femme (et d’autres cellules de son corps) contiennent à la fois des mitochondries normales et mutantes, et que certaines n’ont que quelques mitochondries mutantes, tandis que d’autres en ont beaucoup. Un enfant conçu à partir d’un ovule « principalement sain” ne développerait probablement pas de maladie, et un enfant conçu à partir d’un ovule « principalement mutant” le fera probablement. En outre, la femme peut ou non avoir des symptômes de la maladie mitochondriale elle-même.
ces maladies peuvent également survenir de façon sporadique, ce qui signifie qu’elles peuvent survenir sans antécédents familiaux.,
le risque de transmettre une maladie mitochondriale aux enfants dépend de nombreux facteurs, notamment si la maladie est causée par des mutations de l’adnn ou de l’ADNmt. Un bon moyen d’en savoir plus sur ces risques est de parler à un médecin ou à un conseiller génétique dans un centre de soins MDA. Aussi, voir faits sur la génétique et les maladies neuromusculaires.
Leave a Reply