Causas/Herencia
¿Qué causa las enfermedades mitocondriales?
las miopatías mitocondriales son relativamente comunes. Los trastornos mitocondriales primarios son los errores hereditarios más comunes del metabolismo. La prevalencia de encefalomiopatías mitocondriales en niños en edad preescolar es de 1 en 11.000. La enfermedad mitocondrial causada por mutaciones en el ADN mitocondrial tiene una prevalencia estimada de 1 en 5.000. Sin embargo, la enfermedad mitocondrial causada por mutaciones en el ADN nuclear tiene una prevalencia estimada de 1 en 35.000.,1
Las enfermedades mitocondriales no son contagiosas y no son causadas por nada que una persona haga. Son causadas por mutaciones, o cambios, en los genes — los planos de las células para producir proteínas.
Los Genes son responsables de construir nuestro cuerpo y se transmiten de padres a hijos, junto con cualquier mutación o defecto que tengan. Eso significa que las enfermedades mitocondriales son hereditarias, aunque a menudo afectan a miembros de la misma familia de diferentes maneras. (Para obtener más información sobre las mutaciones genéticas y la enfermedad mitocondrial, consulte a continuación.,)
los genes involucrados en la enfermedad mitocondrial normalmente producen proteínas que funcionan en o sobre las mitocondrias. Dentro de cada mitocondria (la forma singular de mitocondria), estas proteínas forman parte de una línea de ensamblaje que utiliza moléculas de combustible derivadas de los alimentos para fabricar la molécula de energía trifosfato de adenosina (ATP) a través de un proceso llamado fosforilación oxidativa. Este proceso de fabricación altamente eficiente requiere oxígeno; fuera de la mitocondria, hay formas menos eficientes de producir ATP sin oxígeno.,
Las proteínas al comienzo de la línea de ensamblaje mitocondrial actúan como manipuladores de carga, importando las moléculas de combustible — azúcares y grasas — a la mitocondria. Luego, otras proteínas descomponen los azúcares y las grasas, extrayendo energía en forma de partículas cargadas llamadas electrones.
Las proteínas hacia el final de la línea — organizadas en cinco grupos llamados complejos I, II, III, IV y V, y dos portadores móviles de electrones, la coenzima Q10 y el citocromo c — aprovechan la energía de esos electrones para producir ATP., Los complejos I A IV lanzan los electrones por la línea y, por lo tanto, se llaman cadena de transporte de electrones, y el complejo V en realidad produce ATP, por lo que también se llama ATP sintasa.
Una deficiencia en uno o más de estos complejos es la causa típica de una enfermedad mitocondrial. (De hecho, las enfermedades mitocondriales a veces se nombran por una deficiencia específica,como la deficiencia del complejo I.) La deficiencia de coenzima Q10 debido a mutaciones nucleares en el ADN puede presentarse con debilidad muscular proximal de forma aislada., Para funcionar correctamente, las proteínas de la vía de fosforilación oxidativa deben ser traducidas, importadas a las mitocondrias e insertadas en la membrana mitocondrial interna. Las mutaciones en los genes que afectan estos procesos también pueden causar miopatía mitocondrial.
Los defectos en los genes que están relacionados con la estructura y la dinámica de las mitocondrias también pueden estar involucrados en el desarrollo de miopatías., Cuando una célula está llena de mitocondrias defectuosas, no solo se priva de ATP, sino que también puede acumular una acumulación de moléculas de combustible y oxígeno no utilizados, con efectos potencialmente desastrosos.
en tales casos, el exceso de moléculas de combustible se utiliza para producir ATP por medios ineficientes, lo que puede generar subproductos potencialmente dañinos como el ácido láctico. (Esto también ocurre cuando una célula tiene un suministro inadecuado de oxígeno, que puede suceder a las células musculares durante el ejercicio extenuante.,) La acumulación de ácido láctico en la sangre – llamada acidosis láctica – está asociada con la fatiga muscular y en realidad podría dañar el tejido muscular y nervioso.
mientras tanto, el oxígeno no utilizado en la célula se puede convertir en compuestos destructivos llamados especies reactivas de oxígeno, incluidos los llamados radicales libres. (Estos son los objetivos de los medicamentos antioxidantes y vitaminas.)
el ATP derivado de las mitocondrias proporciona la principal fuente de energía para la contracción de las células musculares y la activación de las células nerviosas. Por lo tanto, las células musculares y las células nerviosas son especialmente sensibles a los defectos mitocondriales., Los efectos combinados de la privación de energía y la acumulación de toxinas en estas células probablemente dan lugar a los principales síntomas de miopatías y encefalomiopatías mitocondriales.
En resumen, dependiendo del defecto genético primario, las enfermedades mitocondriales pueden ser causadas por alteraciones de lo siguiente: proteínas de la cadena respiratoria, proteínas auxiliares de la cadena respiratoria, traducción del ARN mitocondrial, entorno lipídico de la membrana interna mitocondrial, agotamiento del ADN mitocondrial y dinámica mitocondrial., Las miopatías mitocondriales también se pueden dividir en categorías en función de cuál de estos procesos se altera.
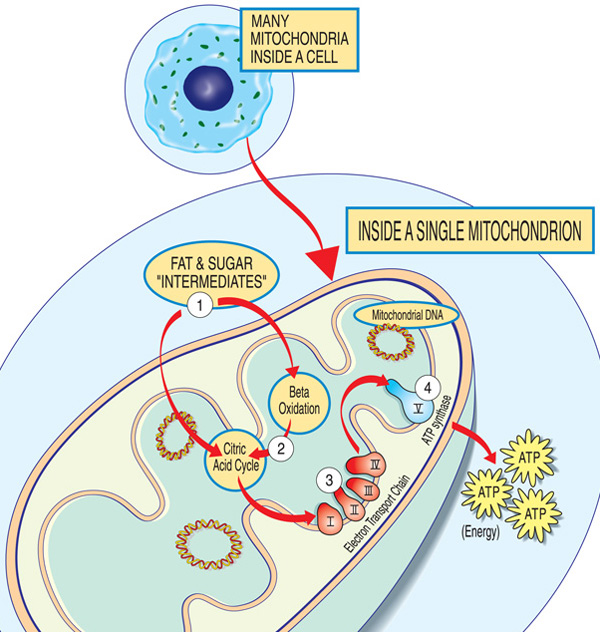
Cada mitocondria es una fábrica de energía que» importa «azúcares y grasas, los descompone y» exporta » energía (ATP) a través de estos pasos: los intermedios de grasa y azúcar ingresan a la mitocondria. Los ácidos grasos se descomponen a través de la oxidación beta y la eliminación de electrones en el ciclo del ácido cítrico. Los electrones pasan a través del complejo principal de la cadena de transporte de electrones. ATP es producido por ATP sintasa.,
¿cuáles son los patrones de herencia en las miopatías mitocondriales?
la genética mitocondrial es compleja y, a menudo, una enfermedad mitocondrial puede ser difícil de rastrear a través de un árbol genealógico. Pero debido a que son causadas por genes defectuosos, las enfermedades mitocondriales son hereditarias.
para entender cómo las enfermedades mitocondriales se transmiten de padres a hijos, es importante saber que hay dos tipos de genes esenciales para las mitocondrias. El primer tipo se encuentra dentro del núcleo — la parte de nuestras células que contiene la mayor parte de nuestro material genético, o ADN., El segundo tipo reside exclusivamente dentro del ADN contenido dentro de las propias mitocondrias.
Las mutaciones en el ADN nuclear (adnn) o en el ADN mitocondrial (ADNmt) pueden causar enfermedad mitocondrial.
La mayor parte del nDNA (junto con cualquier mutación que tenga) se hereda en un patrón mendeliano, lo que significa vagamente que una copia de cada gen proviene de cada padre. Además, la mayoría de las enfermedades mitocondriales causadas por mutaciones en el nDNA (incluyendo el síndrome de Leigh, MNGIE e incluso MDS) son autosómicas recesivas, lo que significa que se necesitan mutaciones en ambas copias de un gen para causar la enfermedad.,
a diferencia del adnn, el ADNmt pasa solo de madre a hijo. Esto se debe a que durante la concepción, cuando el espermatozoide se fusiona con el óvulo, las mitocondrias del espermatozoide — y su ADNmt — se destruyen. Por lo tanto, las enfermedades mitocondriales causadas por mutaciones en el ADNmt son únicas porque se heredan en un patrón materno (ver ilustración a continuación).
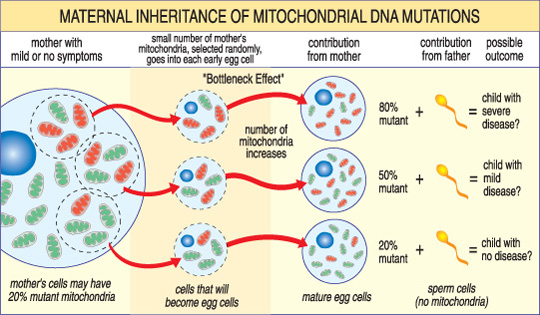
La gravedad de una enfermedad mitocondrial en un niño depende del porcentaje de mitocondrias anormales (mutantes) en el óvulo que las formó.,
otra característica única de las enfermedades del ADNmt surge del hecho de que una célula humana típica — incluyendo el óvulo — contiene solo un núcleo pero cientos de mitocondrias. Una sola célula puede contener mitocondrias mutantes y mitocondrias normales, y el equilibrio entre las dos determinará la salud de la célula. Esto ayuda a explicar por qué los síntomas de la enfermedad mitocondrial pueden variar tanto de una persona a otra, incluso dentro de la misma familia.,
Imagine que los óvulos de una mujer (y otras células en su cuerpo) contienen mitocondrias normales y mutantes, y que algunas tienen solo unas pocas mitocondrias mutantes, mientras que otras tienen muchas. Un niño concebido a partir de un óvulo «en su mayoría sano» probablemente no desarrollaría la enfermedad, y un niño concebido a partir de un óvulo «en su mayoría mutante» probablemente lo hará. Además, la mujer puede o no tener síntomas de la enfermedad mitocondrial.
estas enfermedades también pueden surgir de manera esporádica, lo que significa que pueden ocurrir sin antecedentes familiares.,
el riesgo de transmitir una enfermedad mitocondrial a los niños depende de muchos factores, incluso si la enfermedad es causada por mutaciones en el adnn o el ADNmt. Una buena manera de obtener más información sobre estos riesgos es hablar con un médico o un asesor genético en un centro de atención de MDA. También, ver datos sobre la genética y las Enfermedades Neuromusculares.
Leave a Reply