cauze/moștenire
ce cauzează bolile mitocondriale?miopatiile mitocondriale sunt relativ frecvente. Tulburările mitocondriale primare sunt cele mai frecvente erori moștenite ale metabolismului. Prevalența encefalomiopatiei mitocondriale pentru copiii de vârstă preșcolară este de 1 din 11.000. Boala mitocondrială cauzată de mutații în ADN-ul mitocondrial are o prevalență estimată de 1 din 5000. Cu toate acestea, boala mitocondrială cauzată de mutații în ADN-ul nuclear are o prevalență estimată de 1 în 35.000.,1
bolile mitocondriale nu sunt contagioase și nu sunt cauzate de nimic pe care o face o persoană. Sunt cauzate de mutații sau modificări ale genelor-planurile celulelor pentru producerea proteinelor.genele sunt responsabile pentru construirea corpului nostru și sunt transmise de la părinți la copii, împreună cu orice mutații sau defecte pe care le au. Aceasta înseamnă că bolile mitocondriale sunt moștenite, deși afectează adesea membrii aceleiași familii în moduri diferite. (Pentru mai multe informații despre mutațiile genetice și boala mitocondrială, vezi mai jos.,genele implicate în boala mitocondrială produc în mod normal proteine care funcționează în sau pe mitocondrii. În fiecare mitocondrie (singular formă de mitocondrii), aceste proteine face parte dintr-o linie de asamblare care utilizează combustibil molecule derivate din alimente pentru producerea de energie molecula de adenozină trifosfat (ATP) printr-un proces numit fosforilare oxidativă. Acest proces de fabricație extrem de eficient necesită oxigen; în afara mitocondriilor, există modalități mai puțin eficiente de a produce ATP fără oxigen.,proteinele de la începutul liniei de asamblare mitocondrială acționează ca manipulatorii de marfă, importând moleculele de combustibil — zaharuri și grăsimi — în mitocondrii. Apoi, alte proteine descompun zaharurile și grăsimile, extragând energie sub formă de particule încărcate numite electroni.proteinele spre capătul liniei-organizate în cinci grupe numite complexe I, II, III, IV și V și doi purtători de electroni mobili, coenzima Q10 și citocromul c — valorifică energia acestor electroni pentru a produce ATP., Complexele i prin IV transferă electronii pe linie și, prin urmare, sunt numiți lanțul de transport al electronilor, iar complexul V generează de fapt ATP, deci se mai numește și ATP sintază.o deficiență în unul sau mai multe dintre aceste complexe este cauza tipică a unei boli mitocondriale. (De fapt, bolile mitocondriale sunt uneori numite pentru o deficiență specifică, cum ar fi deficiența complexă I.) Deficiența de coenzima Q10 datorată mutațiilor ADN nucleare poate prezenta slăbiciune musculară proximală în izolare., Pentru a funcționa corect, proteinele căii de fosforilare oxidativă trebuie traduse, importate în mitocondrii și introduse în membrana mitocondrială interioară. Mutațiile genelor care afectează aceste procese pot provoca, de asemenea, miopatie mitocondrială.defectele genelor care sunt legate de structura și dinamica mitocondriilor pot fi, de asemenea, implicate în dezvoltarea miopatiilor., Atunci când o celulă este umplută cu mitocondrii defecte, nu numai că devine lipsită de ATP, ci poate acumula și o întârziere de molecule de combustibil neutilizate și oxigen, cu efecte potențial dezastruoase.în astfel de cazuri, moleculele de combustibil în exces sunt utilizate pentru a face ATP prin mijloace ineficiente, care pot genera produse secundare potențial dăunătoare, cum ar fi acidul lactic. (Acest lucru se întâmplă și atunci când o celulă are o cantitate inadecvată de oxigen, care se poate întâmpla cu celulele musculare în timpul exercițiilor fizice intense.,) Acumularea de acid lactic în sânge — numită acidoză lactică — este asociată cu oboseală musculară și poate afecta efectiv țesutul muscular și nervos.între timp, oxigenul neutilizat din celulă poate fi transformat în compuși distructivi numiți specii reactive de oxigen, inclusiv așa-numiții radicali liberi. (Acestea sunt țintele medicamentelor antioxidante și vitaminelor.ATP derivat din mitocondrii oferă principala sursă de energie pentru contracția celulelor musculare și arderea celulelor nervoase. Deci, celulele musculare și celulele nervoase sunt deosebit de sensibile la defectele mitocondriale., Efectele combinate ale privării de energie și acumulării de toxine în aceste celule dau probabil naștere principalelor simptome ale miopatiilor mitocondriale și encefalomiopatiilor.în rezumat, în funcție de defectul genetic primar, bolile mitocondriale pot fi cauzate de modificări ale următoarelor: proteine ale lanțului respirator, proteine auxiliare ale lanțului respirator, translație ARN mitocondrial, mediu lipidic al membranei interioare mitocondriale, epuizarea ADN-ului mitocondrial și dinamica mitocondrială., Miopatiile mitocondriale pot fi, de asemenea, împărțite în categorii în funcție de care dintre aceste procese este modificat.
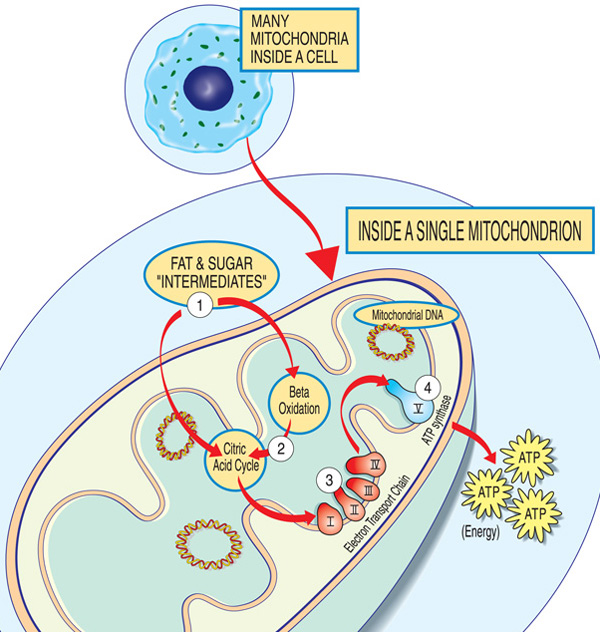
Fiecare mitocondrie este o energie fabrica că „importurile” de zaharuri și grăsimi, le descompune și „exportul” de energie (ATP) prin acești pași: Grăsimi și zahăr intermediari intre mitocondrie. Acizii grași sunt defalcați prin oxidarea beta și îndepărtarea electronilor în ciclul acidului citric. Electronii sunt trecuți prin complexul major al lanțului de transport al electronilor. ATP se face prin ATP synthase.,
care sunt modelele de moștenire în miopatiile mitocondriale?genetica mitocondrială este complexă și, adesea, o boală mitocondrială poate fi dificil de urmărit printr-un arbore genealogic. Dar, deoarece sunt cauzate de gene defecte, bolile mitocondriale se desfășoară în familii.pentru a înțelege modul în care bolile mitocondriale sunt transmise prin familii, este important să știți că există două tipuri de gene esențiale pentru mitocondrii. Primul tip este găzduit în nucleu — partea celulelor noastre care conține cea mai mare parte a materialului nostru genetic sau ADN., Al doilea tip se află exclusiv în ADN-ul conținut în interiorul mitocondriilor.mutațiile fie în ADN-ul nuclear (nDNA), fie în ADN-ul mitocondrial (mtDNA) pot provoca boli mitocondriale.majoritatea nDNA (împreună cu mutațiile pe care le are) este moștenită într-un model Mendelian, ceea ce înseamnă că o copie a fiecărei gene provine de la fiecare părinte. De asemenea, cele mai multe boli mitocondriale cauzate de nDNA mutații (inclusiv sindromul Leigh, MNGIE, și chiar MDS) sunt autozomal recesiv, ceea ce înseamnă că este nevoie de mutații în ambele copii ale unei gene de a provoca boala.,spre deosebire de nDNA, mtDNA trece numai de la mamă la copil. Asta pentru că în timpul concepției, când spermatozoidul se unește cu ovulul, sperma mitocondrii și adn mitocondrial — sunt distruse. Astfel, bolile mitocondriale cauzate de mutațiile mtDNA sunt unice deoarece sunt moștenite într-un model matern (vezi ilustrația de mai jos).
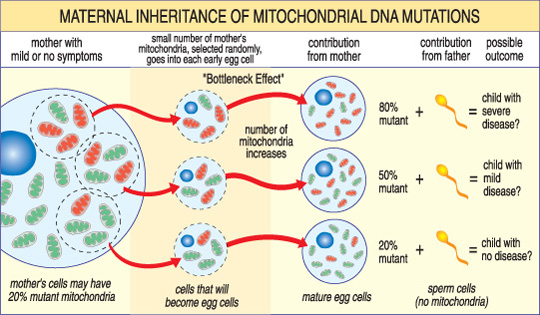
severitatea unei boli mitocondriale la un copil depinde de procentul de anormale (mutant) mitocondriile din celula-ou care le-au format.,o altă caracteristică unică a bolilor mtDNA apare din faptul că o celulă umană tipică — inclusiv celula de ou — conține doar un singur nucleu, dar sute de mitocondrii. O singură celulă poate conține atât mitocondriile mutante, cât și mitocondriile normale, iar echilibrul dintre cele două va determina sănătatea celulei. Acest lucru ajută la explicarea motivului pentru care simptomele bolii mitocondriale pot varia atât de mult de la o persoană la alta, chiar și în cadrul aceleiași familii.,imaginați-vă că celulele ouălor unei femei (și alte celule din corpul ei) conțin atât mitocondrii normale, cât și mutante și că unele au doar câteva mitocondrii mutante, în timp ce altele au multe. Un copil conceput dintr-o celulă de ou” cea mai mare parte sănătoasă „probabil că nu ar dezvolta boală, iar un copil conceput dintr-o celulă de ou” cea mai mare parte mutantă ” probabil o va face. De asemenea, femeia poate sau nu poate avea simptome ale bolii mitocondriale.aceste boli pot apărea, de asemenea, în mod sporadic, ceea ce înseamnă că pot apărea fără antecedente familiale.,riscul de transmitere a unei boli mitocondriale la copii depinde de mulți factori, inclusiv dacă boala este cauzată de mutații în nDNA sau mtDNA. O modalitate bună de a afla mai multe despre aceste riscuri este să discutați cu un medic sau consilier genetic la un centru de îngrijire MDA. De asemenea, vezi fapte despre genetică și boli neuromusculare.
Leave a Reply