causas/herança
quais as causas das doenças mitocondriais?as miopatias mitocondriais são relativamente comuns. As doenças mitocondriais primárias são os erros hereditários mais comuns do metabolismo. A prevalência de encefalomiopatias mitocondriais para crianças em idade pré-escolar é de 1 em 11 000. A doença mitocondrial causada por mutações no ADN mitocondrial tem uma prevalência estimada de 1 em 5.000. No entanto, a doença mitocondrial causada por mutações no DNA nuclear tem uma prevalência estimada de 1 em 35.000.,As doenças mitocondriais não são contagiosas e não são causadas por nada do que uma pessoa faz. São causadas por mutações, ou mudanças, nos genes — as plantas das células para fazer proteínas.os Genes são responsáveis pela construção dos nossos corpos e são passados dos pais para as crianças, juntamente com quaisquer mutações ou defeitos que tenham. Isso significa que as doenças mitocondriais são hereditárias, embora muitas vezes afetem membros da mesma família de maneiras diferentes. (Para mais informação sobre mutações genéticas e doença mitocondrial, ver abaixo.,)
os genes envolvidos na doença mitocondrial normalmente fazem proteínas que funcionam dentro ou sobre a mitocôndria. Dentro de cada mitocôndrio (a forma singular de mitocôndria), estas proteínas compõem parte de uma linha de montagem que usa moléculas de combustível derivadas de alimentos para fabricar a molécula de energia trifosfato de adenosina (ATP) através de um processo chamado fosforilação oxidativa. Este processo de fabricação altamente eficiente requer oxigênio; fora da mitocôndria, há formas menos eficientes de produzir ATP sem oxigênio.,as proteínas no início da linha de montagem mitocondrial agem como tratadores de carga, importando as moléculas de combustível — açúcares e gorduras — para a mitocôndria. Em seguida, outras proteínas quebram os açúcares e gorduras, extraindo energia na forma de partículas carregadas chamadas elétrons.proteínas
para o final da linha-organizado em cinco grupos chamados complexos I, II, III, IV e V, e dois portadores de elétrons móveis, coenzima Q10 e citocromo c — aproveitam a energia desses elétrons para fazer ATP., Complexos I através de IV shuttle os elétrons ao longo da linha e, portanto, são chamados de cadeia de transporte de elétrons, e complexo V Na verdade produz ATP, por isso também é chamado de ATP sintase.uma deficiência em um ou mais destes complexos é a causa típica de uma doença mitocondrial. (Na verdade, as doenças mitocondriais são por vezes nomeadas por uma deficiência específica, como a deficiência complexa I.) A deficiência de coenzima Q10 devido a mutações de ADN nuclear pode apresentar fraqueza muscular proximal isoladamente., Para funcionar correctamente, as proteínas da via de fosforilação oxidativa devem ser traduzidas, importadas para a mitocôndria e inseridas na membrana mitocondrial interna. Mutações em genes que afetam estes processos também podem causar miopatia mitocondrial.defeitos nos genes relacionados com a estrutura e dinâmica da mitocôndria podem também estar envolvidos no desenvolvimento de miopatias., Quando uma célula é preenchida com mitocôndrias defeituosas, não só se torna privada de ATP, também pode acumular um backlog de moléculas de combustível não utilizadas e oxigênio, com efeitos potencialmente desastrosos.em tais casos, as moléculas de combustível em excesso são usadas para produzir ATP por meios ineficientes, que podem gerar subprodutos potencialmente nocivos, como ácido láctico. (Isto também ocorre quando uma célula tem um fornecimento inadequado de oxigênio, o que pode acontecer com as células musculares durante o exercício extenuante.,) O acúmulo de ácido láctico no sangue — denominado acidose láctica — está associado à fadiga muscular e pode, na verdade, danificar o tecido muscular e nervoso.entretanto, o oxigénio não utilizado na célula pode ser convertido em compostos destrutivos chamados espécies reactivas de oxigénio, incluindo os chamados radicais livres. (Estes são os alvos de medicamentos antioxidantes e vitaminas.)
ATP derivado da mitocôndria fornece a principal fonte de energia para a contração das células musculares e o disparo das células nervosas. Portanto, as células musculares e as células nervosas são especialmente sensíveis a defeitos mitocondriais., Os efeitos combinados da privação de energia e acumulação de toxinas nestas células dão provavelmente origem aos principais sintomas das miopatias e encefalomiopatias mitocondriais.
em resumo, dependendo do defeito genético primário, as doenças mitocondriais podem ser causadas por alterações do seguinte: proteínas da cadeia respiratória, proteínas auxiliares da cadeia respiratória, tradução do ARN mitocondrial, meio lipídico da membrana interna mitocondrial, depleção do ADN mitocondrial e dinâmica mitocondrial., As miopatias mitocondriais também podem ser divididas em categorias com base nas quais estes processos são alterados.
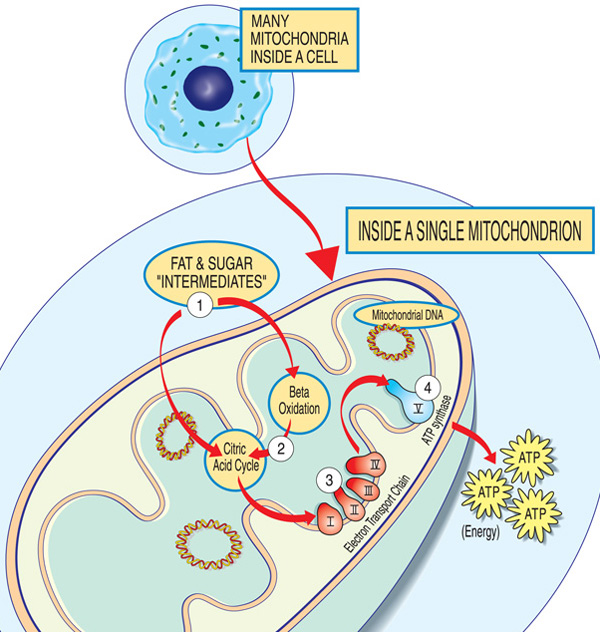
cada mitocôndria é uma fábrica de energia que “importa” açúcares e gorduras, os decompõe e “exporta” energia (ATP) através destes passos: gordura e açúcar intermediários entram na mitocôndria. Os ácidos gordos são decompostos através da oxidação beta e da remoção de electrões no ciclo do ácido cítrico. Os elétrons são passados através do maior complexo da cadeia de transporte de elétrons. A ATP é feita pela ATP synthase.,quais são os padrões de herança nas miopatias mitocondriais?a genética mitocondrial é complexa, e muitas vezes uma doença mitocondrial pode ser difícil de rastrear através de uma árvore genealógica. Mas como são causados por genes defeituosos, as doenças mitocondriais ocorrem em famílias.para compreender como as doenças mitocondriais são transmitidas através das famílias, é importante saber que existem dois tipos de genes essenciais para a mitocôndria. O primeiro tipo é alojado dentro do núcleo — a parte das nossas células que contém a maior parte do nosso material genético, ou ADN., O segundo tipo reside exclusivamente no ADN contido dentro das mitocôndrias propriamente ditas.mutações em ADN nuclear (nDNA) ou ADN mitocondrial (mtDNA) podem causar doença mitocondrial.
a maioria de nDNA (juntamente com quaisquer mutações que tem) é herdada em um padrão Mendeliano, vagamente significando que uma cópia de cada gene vem de cada pai. Além disso, a maioria das doenças mitocondriais causadas por mutações nDNA (incluindo síndrome de Leigh, MNGIE, e até MDS) são autossômicas recessivas, o que significa que são necessárias mutações em ambas as cópias de um gene para causar a doença.,ao contrário de nDNA, mtDNA passa apenas de mãe para filho. Isto porque durante a concepção, quando o esperma se funde com o óvulo, as mitocôndrias do esperma — e o seu mtDNA — são destruídas. Assim, as doenças mitocondriais causadas por mutações mtDNA são únicas porque são herdadas em um padrão materno (ver ilustração abaixo).
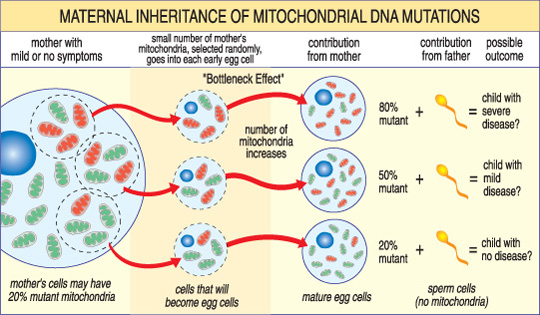
A gravidade de uma doença mitocondrial em uma criança depende da percentagem de anormal (mutante) mitocôndrias do óvulo que os formou.,
outra característica única das doenças mtDNA surge do fato de que uma célula humana típica — incluindo a célula do ovo — contém apenas um núcleo, mas centenas de mitocôndrias. Uma única célula pode conter mitocôndria mutante e mitocôndria normal, e o equilíbrio entre os dois irá determinar a saúde da célula. Isto ajuda a explicar porque os sintomas da doença mitocondrial podem variar tanto de pessoa para pessoa, mesmo dentro da mesma família.,Imagine que os óvulos de uma mulher (e outras células no seu corpo) contêm mitocôndrias normais e mutantes, e que alguns têm apenas algumas mitocôndrias mutantes, enquanto outros têm muitas. Uma criança concebida a partir de uma célula do ovo “em sua maioria saudável” provavelmente não iria desenvolver a doença, e uma criança concebida a partir de uma célula do ovo “em sua maioria mutante” provavelmente irá. Além disso, a mulher pode ou não ter sintomas de doença mitocondrial.estas doenças também podem surgir de forma esporádica, o que significa que podem ocorrer sem história familiar.,
o risco de transmissão de uma doença mitocondrial a crianças depende de muitos factores, incluindo se a doença é causada por mutações em nDNA ou mtDNA. Uma boa maneira de descobrir mais sobre esses riscos é falar com um médico ou conselheiro genético em um centro de cuidados MDA. Veja Também fatos sobre Genética e Doenças Neuromusculares.
Leave a Reply