příčiny / dědičnost
co způsobuje mitochondriální onemocnění?
mitochondriální myopatie jsou poměrně časté. Primární mitochondriální poruchy jsou nejčastějšími zděděnými chybami metabolismu. Prevalence mitochondriálních encefalomyopatií u dětí předškolního věku je 1 z 11 000. Mitochondriální onemocnění způsobené mutacemi v mitochondriální DNA má odhadovanou prevalenci 1 z 5 000. Mitochondriální onemocnění způsobené mutacemi v jaderné DNA má však odhadovanou prevalenci 1 z 35 000.,1
mitochondriální onemocnění nejsou nakažlivá a nejsou způsobena ničím, co člověk dělá. Jsou způsobeny mutacemi nebo změnami v genech-plány buněk pro výrobu proteinů.
geny jsou zodpovědné za budování našeho těla a jsou předávány od rodičů k dětem, spolu s jakýmikoli mutacemi nebo defekty, které mají. To znamená, že mitochondriální onemocnění jsou dědičná, i když často ovlivňují členy stejné rodiny různými způsoby. (Další informace o genetických mutacích a mitochondriálním onemocnění naleznete níže.,)
geny podílející se na mitochondriálním onemocnění obvykle vytvářejí proteiny, které pracují v mitochondriích nebo na nich. V rámci jednotlivých mitochondrií (singulární formu mitochondrie), tyto proteiny tvoří součást montážní linky, který používá palivo molekuly odvozené od potravin k výrobě energie molekuly adenosin trifosfátu (ATP), přes proces s názvem oxidativní fosforylace. Tento vysoce účinný výrobní proces vyžaduje kyslík; mimo mitochondrií existují méně účinné způsoby produkce ATP bez kyslíku.,
Proteiny na začátku mitochondriální montážní linky chovat jako skladníky, dovozu pohonných hmot z molekuly cukrů a tuků do mitochondrií. Další proteiny rozkládají cukry a tuky a extrahují energii ve formě nabitých částic nazývaných elektrony.
Proteiny ke konci line — uspořádány do pěti skupin, tzv. komplexy I, II, III, IV a V, a dva mobilní přenašeče elektronů, koenzym Q10 a cytochrom c — využít energii z těchto elektronů ATP., Komplexy i přes IV shuttle elektrony po linii, a proto se nazývají elektronový transportní řetězec, a komplexní V vlastně chrlí ATP, takže se také nazývá ATP syntáza.
nedostatek v jednom nebo více z těchto komplexů je typickou příčinou mitochondriálního onemocnění. (Ve skutečnosti jsou mitochondriální onemocnění někdy pojmenována pro specifický nedostatek, jako je nedostatek komplexního i.) Nedostatek koenzymu Q10 v důsledku mutací jaderné DNA může být izolován s proximální svalovou slabostí., Aby správně fungovaly, musí být proteiny oxidační fosforylační dráhy přeloženy, dovezeny do mitochondrií a vloženy do vnitřní mitochondriální membrány. Mutace v genech, které ovlivňují tyto procesy, mohou také způsobit mitochondriální myopatii.
defekty genů, které souvisejí se strukturou a dynamikou mitochondrií, se mohou také podílet na vývoji myopatií., Když je buňka naplněna vadnými mitochondriemi, nejenže je zbavena ATP, ale může také akumulovat nevyřízené molekuly nevyužitého paliva a kyslíku s potenciálně katastrofálními účinky.
v takových případech se přebytečné molekuly paliva používají k výrobě ATP neefektivními prostředky, které mohou vytvářet potenciálně škodlivé vedlejší produkty, jako je kyselina mléčná. (K tomu také dochází, když buňka má nedostatečný přívod kyslíku, což se může stát svalovým buňkám během namáhavého cvičení.,) Nahromadění kyseliny mléčné v krvi – nazývané laktátová acidóza-je spojeno se svalovou únavou a může skutečně poškodit svalovou a nervovou tkáň.
mezitím může být nepoužitý kyslík v buňce přeměněn na destruktivní sloučeniny nazývané reaktivní druhy kyslíku, včetně tzv. (To jsou cíle antioxidačních léků a vitamínů.)
ATP odvozený z mitochondrií poskytuje hlavní zdroj energie pro kontrakci svalových buněk a vypalování nervových buněk. Takže svalové buňky a nervové buňky jsou zvláště citlivé na mitochondriální defekty., Kombinované účinky energetické deprivace a akumulace toxinů v těchto buňkách pravděpodobně vedou k hlavním příznakům mitochondriálních myopatií a encefalomyopatií.
stručně řečeno, v závislosti na primární genetický defekt mitochondriální onemocnění může být způsobeno tím, že změny následující: dýchací řetězec bílkovin, dýchací řetězec pomocných proteinů, mitochondriální RNA překlad, vnitřní mitochondriální membrány lipidů prostředí, deplece mitochondriální DNA a mitochondriální dynamiky., Mitochondriální myopatie lze také rozdělit do kategorií na základě toho, který z těchto procesů je změněn.
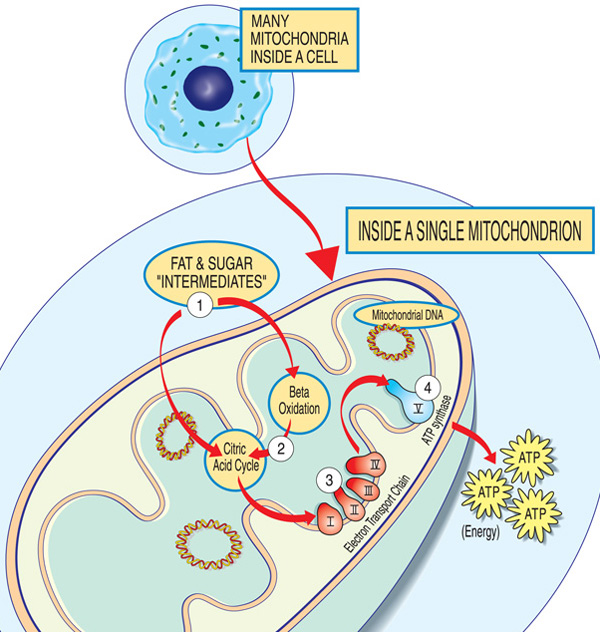
Každá mitochondrie je energetické továrny, že „dovoz“ cukrů a tuků, je láme a „vývoz“ energie (ATP) pomocí těchto kroků: Tuk a cukr meziprodukty vstoupit do mitochondrie. Mastné kyseliny se rozkládají oxidací beta a odstraněním elektronů v cyklu kyseliny citronové. Elektrony procházejí hlavním komplexem elektronového transportního řetězce. ATP je vyroben ATP syntázou.,
jaké jsou dědičné vzorce v mitochondriálních myopatiích?
mitochondriální genetika je složitá a často může být obtížné vysledovat mitochondriální onemocnění rodokmenem. Ale protože jsou způsobeny vadnými geny, mitochondriální onemocnění běží v rodinách.
pochopit, jak mitochondriální onemocnění jsou předávány prostřednictvím rodiny, to je důležité vědět, že existují dva typy geny nezbytné pro mitochondrie. První typ je umístěn v jádru-části našich buněk, která obsahuje většinu našeho genetického materiálu nebo DNA., Druhý typ se nachází výhradně v DNA obsažené uvnitř samotných mitochondrií.
mutace v jaderné DNA (nDNA) nebo mitochondriální DNA (mtDNA) mohou způsobit mitochondriální onemocnění.
Většina nDNA (spolu s mutacemi) je dědičná v Mendelovské vzor, volně, což znamená, že jednu kopii každého genu pochází od každého z rodičů. Také většina mitochondriálních onemocnění způsobených mutacemi nDNA (včetně Leighova syndromu, MNGIE a dokonce i MDS) je autozomálně recesivní, což znamená, že mutace v obou kopiích genu způsobují onemocnění.,
Na rozdíl od nDNA přechází mtDNA pouze z matky na dítě. Je to proto, že během koncepce, kdy se spermie spojí s vejcem, jsou mitochondrie spermií — a její mtDNA — zničeny. Mitochondriální onemocnění způsobená mutacemi mtDNA jsou tedy jedinečná, protože jsou zděděna v mateřském vzoru (viz obrázek níže).
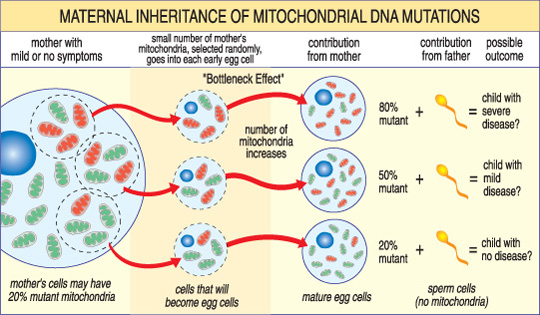
závažnost mitochondriální onemocnění u dítěte závisí na procentu abnormálních (mutant) mitochondrie v buňce vajíčka, která vznikla.,
další jedinečný rys onemocnění mtDNA vyplývá ze skutečnosti, že typická lidská buňka — včetně vaječné buňky — obsahuje pouze jedno jádro, ale stovky mitochondrií. Jedna buňka může obsahovat jak mutantní mitochondrie, tak normální mitochondrie a rovnováha mezi nimi určí zdraví buňky. To pomáhá vysvětlit, proč se příznaky mitochondriální choroby mohou tolik lišit od člověka k člověku, a to i ve stejné rodině.,
Představte si, že ženské vaječné buňky (a další buňky v jejím těle) obsahují normální i mutantní mitochondrie a že některé mají jen několik mutantních mitochondrií, zatímco jiné mají mnoho. Dítě počaté z“ většinou zdravé “ vaječné buňky pravděpodobně nevyvíjí nemoc a dítě počaté z „většinou mutantní“ vaječné buňky pravděpodobně bude. Také žena může nebo nemusí mít příznaky mitochondriální nemoci sama.
tato onemocnění mohou také vzniknout sporadicky, což znamená, že se mohou vyskytnout bez rodinné anamnézy.,
riziko přenosu mitochondriálního onemocnění na děti závisí na mnoha faktorech, včetně toho, zda je onemocnění způsobeno mutacemi v nDNA nebo mtDNA. Dobrým způsobem, jak se dozvědět více o těchto rizicích, je mluvit s lékařem nebo genetickým poradcem v centru péče o MDA. Podívejte se také na fakta o genetice a neuromuskulárních onemocněních.
Leave a Reply