oorzaken/overerving
wat veroorzaakt mitochondriale ziekten?
mitochondriale myopathieën komen relatief vaak voor. Primaire mitochondriale aandoeningen zijn de meest voorkomende erfelijke fouten van het metabolisme. De prevalentie van mitochondriale encefalomyopathieën voor voorschoolse kinderen is 1 op 11.000. Mitochondriale ziekte veroorzaakt door mutaties in mitochondriale DNA heeft een geschatte prevalentie van 1 op 5.000. Echter mitochondriale ziekte veroorzaakt door mutaties in het nucleaire DNA heeft een geschatte prevalentie van 1 op 35.000.,1
mitochondriale ziekten zijn niet besmettelijk en worden niet veroorzaakt door iets wat een persoon doet. Ze worden veroorzaakt door mutaties, of veranderingen, in genen-de blauwdrukken van de cellen voor het maken van eiwitten.
genen zijn verantwoordelijk voor de opbouw van ons lichaam en worden doorgegeven van ouders op kinderen, samen met eventuele mutaties of defecten die ze hebben. Dat betekent dat mitochondriale ziekten erfelijk zijn, hoewel ze vaak leden van dezelfde familie op verschillende manieren beïnvloeden. (Voor meer informatie over genetische mutaties en mitochondriale ziekte, zie hieronder.,)
de genen die betrokken zijn bij mitochondriale aandoeningen maken normaal gesproken eiwitten aan die in of op de mitochondriën werken. Binnen elke mitochondrion (de enkelvoudige vorm van mitochondriën), maken deze proteã nen omhoog deel van een lopende band die brandstofmolecules uit voedsel worden afgeleid gebruikt om de energiemolecule adenosine trifosfaat (ATP) door een proces genoemd oxidatieve phosphorylation te vervaardigen. Dit zeer efficiënte productieproces vereist zuurstof; buiten de mitochondrion, zijn er minder efficiënte manieren om ATP zonder zuurstof te produceren.,
eiwitten aan het begin van de mitochondriale assemblagelijn werken als vrachtafhandelaars en importeren de brandstofmoleculen — suikers en vetten — in het mitochondrion. Vervolgens breken andere eiwitten de suikers en vetten af, waardoor energie wordt gewonnen in de vorm van geladen deeltjes die elektronen worden genoemd.
eiwitten aan het einde van de lijn — georganiseerd in vijf groepen genaamd complexen I, II, III, IV en V, en twee mobiele elektronendragers, co — enzym Q10 en cytochroom c-benutten de energie van die elektronen om ATP te maken., Complexen I tot IV brengen de elektronen langs de lijn en worden daarom de elektronentransportketen genoemd, en complex V maakt eigenlijk ATP, dus het wordt ook wel ATP synthase genoemd.
een deficiëntie in een of meer van deze complexen is de typische oorzaak van een mitochondriale ziekte. (In feite worden mitochondriale ziekten soms genoemd naar een specifieke deficiëntie, zoals complexe I-deficiëntie.) Co-enzym Q10-deficiëntie als gevolg van nucleaire DNA-mutaties kan zich voordoen met proximale spierzwakte in isolatie., Om correct te functioneren, moeten de proteã nen van de oxydatieve phosphorylationweg worden vertaald, in mitochondria worden ingevoerd, en in het binnenste mitochondrial membraan worden ingevoegd. Mutaties in genen die deze processen beïnvloeden kunnen ook mitochondriale myopathie veroorzaken.
defecten in genen die gerelateerd zijn aan de structuur en dynamiek van de mitochondriën kunnen ook betrokken zijn bij de ontwikkeling van myopathieën., Wanneer een cel is gevuld met defecte mitochondriën, niet alleen wordt het beroofd van ATP, het kan ook een achterstand van ongebruikte brandstofmoleculen en zuurstof accumuleren, met potentieel rampzalige gevolgen.
in dergelijke gevallen worden overtollige brandstofmoleculen gebruikt om ATP op inefficiënte wijze aan te maken, wat potentieel schadelijke bijproducten zoals melkzuur kan genereren. (Dit gebeurt ook wanneer een cel een onvoldoende zuurstoftoevoer heeft, wat kan gebeuren met spiercellen tijdens zware inspanning.,) De ophoping van melkzuur in het bloed — lactaatacidose genoemd — wordt geassocieerd met spiervermoeidheid en kan daadwerkelijk spier-en zenuwweefsel beschadigen.
ondertussen kan ongebruikte zuurstof in de cel worden omgezet in destructieve verbindingen die reactieve zuurstofsoorten worden genoemd, inclusief zogenaamde vrije radicalen. (Dit zijn de doelwitten van antioxidanten en vitamines.)
ATP afgeleid van mitochondriën is de belangrijkste bron van kracht voor spiercelcontractie en zenuwcelvuur. Spiercellen en zenuwcellen zijn dus bijzonder gevoelig voor mitochondriale defecten., De gecombineerde effecten van energiegebrek en toxineaccumulatie in deze cellen geven waarschijnlijk aanleiding tot de belangrijkste symptomen van mitochondriale myopathieën en encefalomyopathieën.
samengevat, afhankelijk van het primaire genetische defect, kunnen mitochondriale ziekten worden veroorzaakt door veranderingen van de volgende: respiratoire keten eiwitten, respiratoire keten ondersteunende eiwitten, mitochondriale RNA vertaling, mitochondriale inner membraan lipide milieu, depletie van mitochondriale DNA, en mitochondriale dynamiek., Mitochondriale myopathieën kunnen ook worden onderverdeeld in categorieën gebaseerd op welke van deze processen wordt gewijzigd.
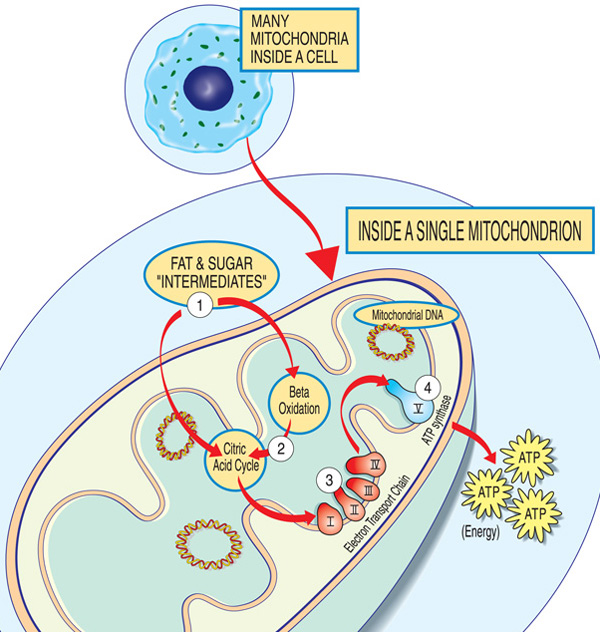
elke mitochondrion is een energiefabriek die suikers en vetten” importeert”, deze opsplitst en energie (ATP) exporteert via deze stappen: tussenproducten van vet en suiker komen in de mitochondrion terecht. Vetzuren worden afgebroken door bèta-oxidatie en de verwijdering van elektronen in de citroenzuurcyclus. Elektronen worden doorgegeven door het belangrijkste complex van de elektronentransportketen. ATP wordt gemaakt door ATP synthase.,
Wat zijn de overervingspatronen bij mitochondriale myopathieën?
mitochondriale genetica zijn complex en vaak kan een mitochondriale ziekte moeilijk te traceren zijn via een stamboom. Maar omdat ze worden veroorzaakt door gebrekkige genen, komen mitochondriale ziekten in families voor.
om te begrijpen hoe mitochondriale ziekten via families worden doorgegeven, is het belangrijk om te weten dat er twee soorten genen zijn die essentieel zijn voor mitochondriën. Het eerste type bevindt zich in de kern — het deel van onze cellen dat het grootste deel van ons genetisch materiaal of DNA bevat., Het tweede type verblijft uitsluitend binnen DNA dat binnen de mitochondria zelf is vervat.
mutaties in nucleair DNA (nDNA) of mitochondriaal DNA (mtDNA) kunnen mitochondriale ziekte veroorzaken.
De meeste ndna (samen met alle mutaties die het heeft) wordt overgeërfd in een Mendeliaans patroon, wat losjes betekent dat een kopie van elk gen afkomstig is van elke ouder. Ook zijn de meeste mitochondriale ziekten veroorzaakt door ndna-mutaties (met inbegrip van het syndroom van Leigh, MNGIE, en zelfs MDS) autosomaal recessief, wat betekent dat het mutaties in beide kopieën van een gen nodig heeft om ziekte te veroorzaken.,
in tegenstelling tot nDNA gaat mtDNA alleen over van moeder op kind. Dat komt omdat tijdens de conceptie, wanneer het sperma fuseert met het ei, de mitochondria van het sperma — en zijn mtDNA — worden vernietigd. Aldus, zijn de mitochondrial ziekten die door mtDNA-veranderingen worden veroorzaakt uniek omdat zij in een moederpatroon worden geërfd (zie illustratie hieronder).
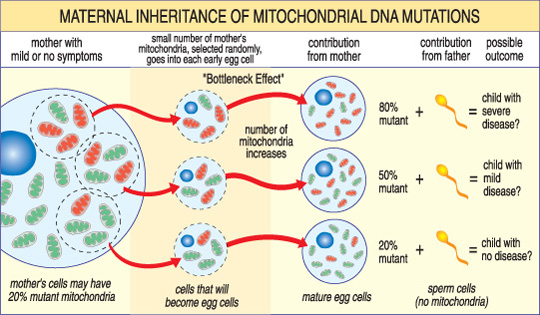
de ernst van een mitochondriale ziekte bij een kind hangt af van het percentage abnormale (mutante) mitochondriën in de eicel die ze vormden.,
een ander uniek kenmerk van mtDNA — ziekten komt voort uit het feit dat een typische menselijke cel — met inbegrip van de eicel-slechts één kern maar honderden mitochondriën bevat. Een enkele cel kan zowel mutante mitochondriën en normale mitochondriën bevatten, en het evenwicht tussen de twee zal de gezondheid van de cel bepalen. Dit helpt verklaren waarom de symptomen van mitochondriale ziekte kan zo veel variëren van persoon tot persoon, zelfs binnen dezelfde familie.,stel je voor dat de eicellen van een vrouw (en andere cellen in haar lichaam) zowel normale als gemuteerde mitochondriën bevatten, en dat sommige slechts een paar gemuteerde mitochondriën hebben, terwijl anderen er veel hebben. Een kind verwekt uit een” meestal gezonde “eicel zou waarschijnlijk geen ziekte ontwikkelen, en een kind verwekt uit een” meestal mutant ” eicel waarschijnlijk wel. Ook kan de vrouw al dan niet symptomen van mitochondriale ziekte zelf.
deze ziekten kunnen ook sporadisch voorkomen, wat betekent dat ze zonder familiegeschiedenis kunnen voorkomen.,
het risico op overdracht van een mitochondriale ziekte op kinderen hangt af van vele factoren, waaronder of de ziekte wordt veroorzaakt door mutaties in nDNA of mtDNA. Een goede manier om meer te weten te komen over deze risico ‘ s is om te praten met een arts of genetische counselor bij een MDA Care Center. Zie ook feiten over genetica en neuromusculaire ziekten.
Leave a Reply