Ursachen/Vererbung
Was verursacht mitochondriale Erkrankungen?
Mitochondriale Myopathien sind relativ häufig. Primäre mitochondriale Störungen sind die häufigsten vererbten Stoffwechselstörungen. Die Prävalenz von mitochondrialen Enzephalomyopathien bei Kindern im Vorschulalter beträgt 1 von 11.000. Mitochondriale Erkrankungen, die durch Mutationen in mitochondrialer DNA verursacht werden, haben eine geschätzte Prävalenz von 1 zu 5.000. Mitochondriale Erkrankungen, die durch Mutationen in der Kern-DNA verursacht werden, haben jedoch eine geschätzte Prävalenz von 1 zu 35.000.,1
Mitochondriale Erkrankungen sind nicht ansteckend und werden durch nichts verursacht, was eine Person tut. Sie werden durch Mutationen oder Veränderungen in Genen verursacht-die Blaupausen der Zellen für die Herstellung von Proteinen.
Gene sind verantwortlich für den Aufbau unseres Körpers und werden von den Eltern an Kinder weitergegeben, zusammen mit Mutationen oder Defekten, die sie haben. Das bedeutet, dass mitochondriale Erkrankungen vererbbar sind, obwohl sie häufig Mitglieder derselben Familie auf unterschiedliche Weise betreffen. (Weitere Informationen zu genetischen Mutationen und mitochondrialen Erkrankungen finden Sie unten.,)
Die Gene, die an mitochondrialen Erkrankungen beteiligt sind, bilden normalerweise Proteine, die in oder auf den Mitochondrien wirken. Innerhalb jedes Mitochondriums (der singulären Form der Mitochondrien) bilden diese Proteine einen Teil einer Montagelinie, die Kraftstoffmoleküle aus der Nahrung verwendet, um das Energiemolekül Adenosintriphosphat (ATP) durch einen Prozess namens oxidative Phosphorylierung herzustellen. Dieser hocheffiziente Herstellungsprozess erfordert Sauerstoff; Außerhalb des Mitochondriums gibt es weniger effiziente Möglichkeiten, ATP ohne Sauerstoff zu produzieren.,
Proteine am Anfang der mitochondrialen Montagelinie wirken wie Frachthandler und importieren die Kraftstoffmoleküle-Zucker und Fette — in das Mitochondrium. Als nächstes brechen andere Proteine die Zucker und Fette ab und extrahieren Energie in Form geladener Teilchen, die Elektronen genannt werden.
Proteine gegen Ende der Linie — organisiert in fünf Gruppen, die Komplexe I, II, III, IV und V genannt werden, und zwei mobile Elektronenträger, Coenzym Q10 und Cytochrom c — nutzen die Energie dieser Elektronen, um ATP herzustellen., Die Komplexe I bis IV pendeln die Elektronen entlang der Linie und werden daher als Elektronentransportkette bezeichnet, und der Komplex V erzeugt tatsächlich ATP, so dass er auch als ATP-Synthase bezeichnet wird.
Ein Mangel an einem oder mehreren dieser Komplexe ist die typische Ursache einer mitochondrialen Erkrankung. (Tatsächlich werden mitochondriale Erkrankungen manchmal nach einem bestimmten Mangel wie Komplex-I-Mangel benannt.) Coenzym – Q10-Mangel aufgrund nuklearer DNA-Mutationen kann isoliert mit proximaler Muskelschwäche auftreten., Um richtig zu funktionieren, müssen die Proteine des oxidativen Phosphorylierungswegs übersetzt, in die Mitochondrien importiert und in die innere Mitochondrienmembran eingeführt werden. Mutationen in Genen, die diese Prozesse beeinflussen, können auch mitochondriale Myopathie verursachen.
Defekte in Genen, die mit der Struktur und Dynamik der Mitochondrien zusammenhängen, können auch an der Entwicklung von Myopathien beteiligt sein., Wenn eine Zelle mit defekten Mitochondrien gefüllt ist, wird ihr nicht nur ATP entzogen, sondern sie kann auch einen Rückstand ungenutzter Brennstoffmoleküle und Sauerstoff mit potenziell katastrophalen Auswirkungen ansammeln.
In solchen Fällen werden überschüssige Kraftstoffmoleküle verwendet, um ATP mit ineffizienten Mitteln herzustellen, die potenziell schädliche Nebenprodukte wie Milchsäure erzeugen können. (Dies tritt auch auf, wenn eine Zelle eine unzureichende Sauerstoffversorgung hat, was Muskelzellen während anstrengenden Trainings passieren kann.,) Der Aufbau von Milchsäure im Blut – Laktatazidose genannt-ist mit Muskelermüdung verbunden und kann tatsächlich Muskel-und Nervengewebe schädigen.
In der Zwischenzeit kann ungenutzter Sauerstoff in der Zelle in destruktive Verbindungen umgewandelt werden, die als reaktive Sauerstoffspezies bezeichnet werden, einschließlich sogenannter freier Radikale. (Dies sind die Ziele von Antioxidantien und Vitaminen.)
ATP, das aus Mitochondrien gewonnen wird, stellt die Hauptenergiequelle für Muskelzellkontraktion und Nervenzellfeuerung dar. So sind Muskelzellen und Nervenzellen besonders empfindlich gegenüber mitochondrialen Defekten., Die kombinierten Auswirkungen von Energieentzug und Toxinansammlung in diesen Zellen führen wahrscheinlich zu den Hauptsymptomen von mitochondrialen Myopathien und Enzephalomyopathien.
Zusammenfassend können mitochondriale Erkrankungen je nach primärem genetischen Defekt durch Veränderungen der folgenden Faktoren verursacht werden: Atmungskettenproteine, Hilfsproteine der Atmungskette, mitochondriale RNA-Translation, mitochondriales Lipidmilieu der inneren Membran, Erschöpfung der mitochondrialen DNA und mitochondriale Dynamik., Mitochondriale Myopathien können auch in Kategorien unterteilt werden, je nachdem, welcher dieser Prozesse verändert ist.
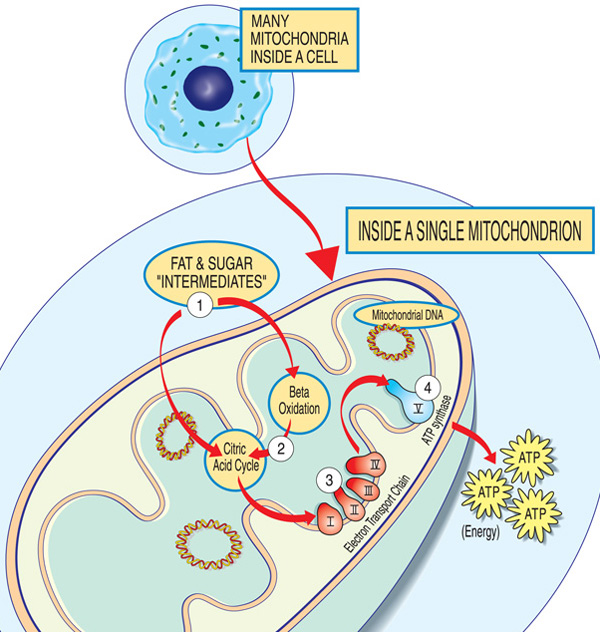
Jedes Mitochondrium ist eine Energiefabrik, die Zucker und Fette“ importiert“, abbaut und“ exportiert “ Energie (ATP) über diese Schritte: Fett und Zuckerzwischenprodukte gelangen in das Mitochondrium. Fettsäuren werden durch Beta-Oxidation und die Entfernung von Elektronen im Zitronensäurekreislauf abgebaut. Elektronen werden durch den Hauptkomplex der Elektronentransportkette geleitet. ATP wird durch ATP-Synthase hergestellt.,
Was sind die Vererbung Muster in der mitochondrialen myopathien?
der Mitochondrialen Genetik sind Komplex, und oft ist eine Mitochondrien-Erkrankung kann schwierig sein zu verfolgen, die durch einen Stammbaum. Aber weil sie durch defekte Gene verursacht werden,laufen mitochondriale Erkrankungen in Familien.
Um zu verstehen, wie mitochondriale Erkrankungen durch Familien weitergegeben werden, ist es wichtig zu wissen, dass es zwei Arten von Genen gibt, die für Mitochondrien essentiell sind. Der erste Typ befindet sich im Kern — dem Teil unserer Zellen, der den größten Teil unseres genetischen Materials oder unserer DNA enthält., Der zweite Typ befindet sich ausschließlich in der DNA, die in den Mitochondrien selbst enthalten ist.
Mutationen in nuklearer DNA (nDNA) oder mitochondrialer DNA (mtDNA) können mitochondriale Erkrankungen verursachen.
Die meisten nDNA (zusammen mit allen Mutationen) werden in einem mendelschen Muster vererbt, was lose bedeutet, dass eine Kopie jedes Gens von jedem Elternteil stammt. Außerdem sind die meisten mitochondrialen Erkrankungen, die durch nDNA-Mutationen verursacht werden (einschließlich Leigh-Syndrom, MNGIE und sogar MDS), autosomal rezessiv, was bedeutet, dass Mutationen in beiden Kopien eines Gens erforderlich sind, um Krankheiten zu verursachen.,
Im Gegensatz zu nDNA geht mtDNA nur von Mutter zu Kind über. Das liegt daran, dass während der Empfängnis, wenn das Sperma mit dem Ei verschmilzt, die Mitochondrien des Spermas — und seine mtDNA — zerstört werden. Daher sind mitochondriale Erkrankungen, die durch mtDNA-Mutationen verursacht werden, einzigartig, da sie mütterlicherseits vererbt werden (siehe Abbildung unten).
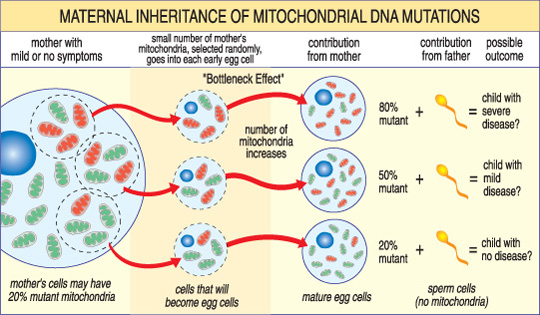
Der Schweregrad einer mitochondrialen Erkrankung bei einem Kind hängt vom Prozentsatz abnormaler (mutierter) Mitochondrien in der Eizelle ab, die sie gebildet hat.,
Ein weiteres einzigartiges Merkmal von mtDNA-Erkrankungen ergibt sich aus der Tatsache, dass eine typische menschliche Zelle — einschließlich der Eizelle — nur einen Kern, aber Hunderte von Mitochondrien enthält. Eine einzelne Zelle kann sowohl mutierte Mitochondrien als auch normale Mitochondrien enthalten, und das Gleichgewicht zwischen den beiden bestimmt die Gesundheit der Zelle. Dies hilft zu erklären, warum die Symptome einer mitochondrialen Erkrankung von Person zu Person so stark variieren können, sogar innerhalb derselben Familie.,
Stellen Sie sich vor, dass die Eizellen einer Frau (und andere Zellen in ihrem Körper) sowohl normale als auch mutierte Mitochondrien enthalten und dass einige nur wenige mutierte Mitochondrien haben, während andere viele haben. Ein Kind, das aus einer „meist gesunden“ Eizelle gezeugt wurde, würde wahrscheinlich keine Krankheit entwickeln, und ein Kind, das aus einer „meist mutierten“ Eizelle gezeugt wurde, wird es wahrscheinlich tun. Auch kann die Frau selbst Symptome einer mitochondrialen Erkrankung haben oder nicht.
Diese Krankheiten können auch sporadisch auftreten, was bedeutet, dass sie ohne Familienanamnese auftreten können.,
Das Risiko, eine mitochondriale Erkrankung an Kinder weiterzugeben, hängt von vielen Faktoren ab, einschließlich der Frage, ob die Krankheit durch Mutationen in nDNA oder mtDNA verursacht wird. Eine gute Möglichkeit, mehr über diese Risiken zu erfahren, besteht darin, mit einem Arzt oder genetischen Berater in einem MDA-Versorgungszentrum zu sprechen. Siehe auch Fakten über Genetik und neuromuskuläre Erkrankungen.
Leave a Reply