Cause/Ereditarietà
Quali sono le cause delle malattie mitocondriali?
Le miopatie mitocondriali sono relativamente comuni. I disturbi mitocondriali primari sono gli errori ereditari più comuni del metabolismo. La prevalenza di encefalomiopatie mitocondriali per i bambini in età prescolare è di 1 su 11.000. La malattia mitocondriale causata da mutazioni nel DNA mitocondriale ha una prevalenza stimata di 1 su 5.000. Tuttavia la malattia mitocondriale causata da mutazioni nel DNA nucleare ha una prevalenza stimata di 1 su 35.000.,1
Le malattie mitocondriali non sono contagiose e non sono causate da nulla che una persona faccia. Sono causati da mutazioni, o cambiamenti, nei geni-i progetti delle cellule per produrre proteine.
I geni sono responsabili della costruzione del nostro corpo e vengono trasmessi dai genitori ai figli, insieme a qualsiasi mutazione o difetto che hanno. Ciò significa che le malattie mitocondriali sono ereditabili, anche se spesso colpiscono i membri della stessa famiglia in modi diversi. (Per ulteriori informazioni sulle mutazioni genetiche e sulla malattia mitocondriale, vedi sotto.,)
I geni coinvolti nella malattia mitocondriale normalmente producono proteine che lavorano nei o sui mitocondri. All’interno di ogni mitocondrio (la forma singolare dei mitocondri), queste proteine costituiscono parte di una catena di montaggio che utilizza molecole di carburante derivate dal cibo per produrre la molecola di energia adenosina trifosfato (ATP) attraverso un processo chiamato fosforilazione ossidativa. Questo processo di produzione altamente efficiente richiede ossigeno; al di fuori del mitocondrio, ci sono modi meno efficienti di produrre ATP senza ossigeno.,
Le proteine all’inizio della catena di montaggio mitocondriale agiscono come trasportatori di carico, importando le molecole di carburante — zuccheri e grassi — nel mitocondrio. Successivamente, altre proteine abbattono gli zuccheri e i grassi, estraendo energia sotto forma di particelle cariche chiamate elettroni.
Le proteine verso la fine della linea — organizzate in cinque gruppi chiamati complessi I, II, III, IV e V, e due portatori di elettroni mobili, il coenzima Q10 e il citocromo c — sfruttano l’energia di quegli elettroni per creare ATP., Complessi I attraverso IV navetta gli elettroni lungo la linea e sono quindi chiamati la catena di trasporto di elettroni, e complesso V in realtà sforna ATP, quindi è anche chiamato ATP sintasi.
Una carenza in uno o più di questi complessi è la causa tipica di una malattia mitocondriale. (In effetti, le malattie mitocondriali sono talvolta chiamate per una carenza specifica,come la carenza di complesso I.) La carenza di coenzima Q10 dovuta a mutazioni del DNA nucleare può presentarsi con debolezza muscolare prossimale in isolamento., Per funzionare correttamente, le proteine della via di fosforilazione ossidativa devono essere tradotte, importate nei mitocondri e inserite nella membrana mitocondriale interna. Mutazioni nei geni che influenzano questi processi possono anche causare miopatia mitocondriale.
Difetti nei geni correlati alla struttura e alla dinamica dei mitocondri possono anche essere coinvolti nello sviluppo delle miopatie., Quando una cellula è piena di mitocondri difettosi, non solo diventa priva di ATP, ma può anche accumulare un arretrato di molecole di carburante inutilizzate e ossigeno, con effetti potenzialmente disastrosi.
In questi casi, le molecole di combustibile in eccesso vengono utilizzate per produrre ATP con mezzi inefficienti, che possono generare sottoprodotti potenzialmente dannosi come l’acido lattico. (Questo si verifica anche quando una cellula ha un apporto di ossigeno inadeguato, che può accadere alle cellule muscolari durante un intenso esercizio fisico.,) L’accumulo di acido lattico nel sangue – chiamato acidosi lattica – è associato all’affaticamento muscolare e potrebbe effettivamente danneggiare il tessuto muscolare e nervoso.
Nel frattempo, l’ossigeno inutilizzato nella cellula può essere convertito in composti distruttivi chiamati specie reattive dell’ossigeno, compresi i cosiddetti radicali liberi. (Questi sono gli obiettivi dei farmaci antiossidanti e delle vitamine.)
L’ATP derivato dai mitocondri fornisce la principale fonte di energia per la contrazione delle cellule muscolari e la cottura delle cellule nervose. Quindi, le cellule muscolari e le cellule nervose sono particolarmente sensibili ai difetti mitocondriali., Gli effetti combinati della privazione di energia e dell’accumulo di tossine in queste cellule danno probabilmente origine ai principali sintomi delle miopatie mitocondriali e delle encefalomiopatie.
In sintesi, a seconda del difetto genetico primario, le malattie mitocondriali possono essere causate da alterazioni delle seguenti: proteine della catena respiratoria, proteine accessorie della catena respiratoria, traduzione dell’RNA mitocondriale, milieu lipidico della membrana interna mitocondriale, deplezione del DNA mitocondriale e dinamica mitocondriale., Le miopatie mitocondriali possono anche essere suddivise in categorie in base a quale di questi processi è alterato.
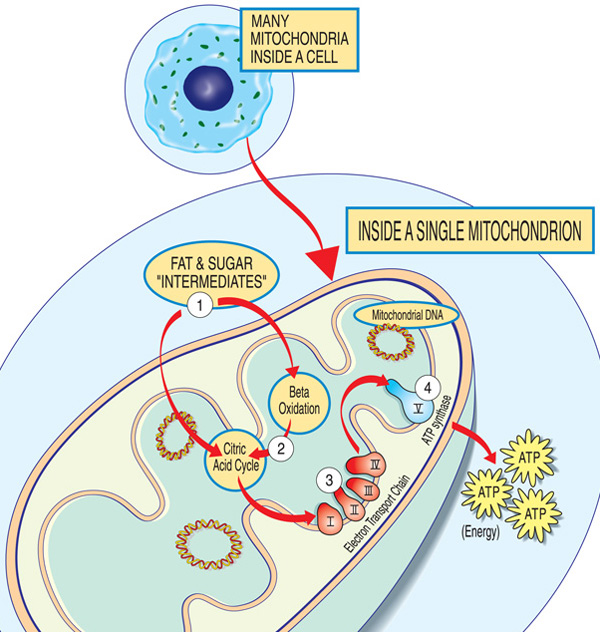
Ogni mitocondrio è una fabbrica di energia che “importa” zuccheri e grassi, li scompone e “esporta” energia (ATP) attraverso questi passaggi: intermedi di grassi e zuccheri entrano nel mitocondrio. Gli acidi grassi vengono scomposti attraverso l’ossidazione beta e la rimozione degli elettroni nel ciclo dell’acido citrico. Gli elettroni sono passati attraverso il complesso principale della catena di trasporto degli elettroni. ATP è fatto da ATP sintasi.,
Quali sono i modelli di ereditarietà nelle miopatie mitocondriali?
La genetica mitocondriale è complessa e spesso una malattia mitocondriale può essere difficile da rintracciare attraverso un albero genealogico. Ma poiché sono causati da geni difettosi, le malattie mitocondriali corrono nelle famiglie.
Per capire come le malattie mitocondriali vengono trasmesse attraverso le famiglie, è importante sapere che ci sono due tipi di geni essenziali per i mitocondri. Il primo tipo è alloggiato all’interno del nucleo — la parte delle nostre cellule che contiene la maggior parte del nostro materiale genetico, o DNA., Il secondo tipo risiede esclusivamente all’interno del DNA contenuto all’interno dei mitocondri stessi.
Mutazioni nel DNA nucleare (nDNA) o nel DNA mitocondriale (mtDNA) possono causare malattie mitocondriali.
La maggior parte degli nDNA (insieme a tutte le mutazioni che ha) è ereditata in un modello mendeliano, il che significa vagamente che una copia di ciascun gene proviene da ciascun genitore. Inoltre, la maggior parte delle malattie mitocondriali causate da mutazioni nDNA (inclusa la sindrome di Leigh, MNGIE e persino MDS) sono autosomiche recessive, il che significa che richiede mutazioni in entrambe le copie di un gene per causare la malattia.,
A differenza di nDNA, mtDNA passa solo da madre a figlio. Questo perché durante il concepimento, quando lo sperma si fonde con l’uovo, i mitocondri dello sperma — e il suo mtDNA — vengono distrutti. Pertanto, le malattie mitocondriali causate da mutazioni mtDNA sono uniche perché sono ereditate in un modello materno (vedi illustrazione sotto).
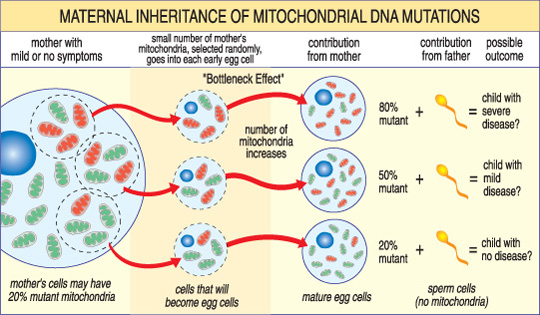
La gravità di una malattia mitocondriale in un bambino dipende dalla percentuale di mitocondri anormali (mutanti) nella cellula uovo che li ha formati.,
Un’altra caratteristica unica delle malattie mtDNA deriva dal fatto che una tipica cellula umana — inclusa la cellula uovo — contiene solo un nucleo ma centinaia di mitocondri. Una singola cellula può contenere sia mitocondri mutanti che mitocondri normali e l’equilibrio tra i due determinerà la salute della cellula. Questo aiuta a spiegare perché i sintomi della malattia mitocondriale possono variare così tanto da persona a persona, anche all’interno della stessa famiglia.,
Immagina che le cellule uovo di una donna (e altre cellule nel suo corpo) contengano mitocondri normali e mutanti, e che alcuni abbiano solo pochi mitocondri mutanti, mentre altri ne hanno molti. Un bambino concepito da una cellula uovo” per lo più sana “probabilmente non svilupperebbe la malattia, e un bambino concepito da una cellula uovo” per lo più mutante” probabilmente lo farà. Inoltre, la donna può o non può avere sintomi della malattia mitocondriale stessa.
Queste malattie possono anche insorgere in modo sporadico, il che significa che possono verificarsi senza storia familiare.,
Il rischio di trasmettere una malattia mitocondriale ai bambini dipende da molti fattori, tra cui se la malattia è causata da mutazioni in nDNA o mtDNA. Un buon modo per saperne di più su questi rischi è quello di parlare con un medico o consulente genetico in un centro di assistenza MDA. Inoltre, vedi Fatti sulla genetica e sulle malattie neuromuscolari.
Leave a Reply