Aiheuttaa/Perintö
Mikä aiheuttaa mitokondrioiden sairauksia?
Mitokondrioiden myopatiat ovat suhteellisen yleisiä. Primaariset mitokondriohäiriöt ovat yleisimpiä aineenvaihdunnan periytyviä virheitä. Alle kouluikäisten lasten mitokondriaalisten enkefalomyopatioiden esiintyvyys on 1: 11 000. Mitokondrion DNA: n mutaatioiden aiheuttaman mitokondriosairauden esiintyvyys on arviolta 1 5 000: sta. Mutaatioiden aiheuttama mitokondriotauti ydin-DNA: ssa on kuitenkin arviolta 1: n 35 000: n esiintyvyys.,1
Mitokondriosairaudet eivät tartu, eivätkä ne johdu mistään, mitä ihminen tekee. Ne johtuvat mutaatioista tai muutoksista geeneissä-solujen piirustuksista proteiinien valmistamiseksi.
geenit ovat vastuussa kehomme rakentamisesta, ja ne siirtyvät vanhemmilta lapsille mahdollisten mutaatioiden tai vikojen mukana. Se tarkoittaa, että mitokondriosairaudet ovat periytyviä, vaikka ne vaikuttavat usein saman perheen jäseniin eri tavoin. (Lisätietoja geneettisistä mutaatioista ja mitokondriotaudista on alla.,)
mitokondriotautiin osallistuvat geenit tekevät normaalisti proteiineja, jotka vaikuttavat mitokondrioissa tai niiden pinnalla. Kussakin mitochondrion (yksikkömuotoa mitokondrioita), nämä proteiinit muodostavat osan kokoonpanolinjalla, joka käyttää polttoaineen molekyylit johdettu elintarvikkeiden valmistus energia molekyyli adenosiini trifosfaatiksi (ATP) kautta prosessi nimeltä oksidatiivinen fosforylaatio. Tämä erittäin tehokas valmistusprosessi vaatii happea; ulkopuolella mitochondrion, on vähemmän tehokkaita tapoja tuottaa ATP: tä ilman happea.,
Proteiinit alussa mitokondrioiden kokoonpanolinjalla toimia, kuten rahdin käsittelijät, tuominen polttoaineen molekyylit — sokerit ja rasvat — osaksi mitochondrion. Seuraava, muut proteiinit hajoavat sokerit ja rasvat talteen energiaa muodossa varautuneita hiukkasia kutsutaan elektroneja.
Proteiinit loppua kohti linja — jaettu viiteen ryhmään kutsutaan komplekseja I, II, III, IV ja V, ja kaksi mobiili electron harjoittajat, koentsyymi Q10 ja sytokromi c — valjaat energiaa ne elektronit tehdä ATP: tä., Kompleksit I-IV shuttle elektronit ruodussa ja ovat siksi kutsutaan elektroninsiirtoketju, ja monimutkainen V todella pystöissä pois ATP, joten sitä kutsutaan myös ATP nopaliinisyntaasin.
yhden tai useamman kompleksin puutos on tyypillinen syy mitokondriotautiin. (Itse asiassa mitokondriosairaudet ovat joskus nimetty tietyn puutteen, kuten monimutkainen I puutos.) Ydin-DNA-mutaatioista johtuva koentsyymi Q10: n puutos voi esiintyä proksimaalisen lihasheikkouden kanssa eristyksessä., Toimiakseen oikein, proteiinit oksidatiivisen fosforylaation koulutusjakson on oltava käännetty, tuotu mitokondrioita, ja työnnetään sisempi mitokondrion kalvo. Näihin prosesseihin vaikuttavat geenien mutaatiot voivat myös aiheuttaa mitokondrioiden myopatiaa.
mitokondrioiden rakenteeseen ja dynamiikkaan liittyvien geenien viat voivat olla mukana myös myopatioiden kehityksessä., Kun solu on täynnä viallisia mitokondrioita, se ei ainoastaan tullut vailla ATP, se voi myös kertyä ruuhkaa käyttämättömän polttoaineen molekyylit ja happea, mahdollisesti tuhoisia vaikutuksia.
tällaisissa tapauksissa, liiallinen polttoaineen molekyylit ovat tottuneet tekemään ATP, jonka tehoton keino, joka voi tuottaa mahdollisesti haitallisia sivutuotteita, kuten maitohappoa. (Tämä tapahtuu myös silloin, kun solulla on riittämätön hapensaanti, joka voi tapahtua lihassoluille rasittavan liikunnan aikana.,) Kertyminen maitohappoa veressä — nimeltään maitohappoasidoosi — liittyy lihasten väsymistä ja saattaa jopa vahingoittaa lihas-ja hermokudoksen.
Samaan aikaan, käyttämätön happi solussa voidaan muuntaa tuhoavia yhdisteitä kutsutaan reaktiivinen hapen lajien, mukaan lukien ns. vapaat radikaalit. (Nämä ovat antioksidanttisten lääkkeiden ja vitamiinien kohteet.)
mitokondrioista johdettu ATP tarjoaa lihassolujen supistumisen ja hermosolulaukauksen pääasiallisen voimanlähteen. Lihassolut ja hermosolut ovat siis erityisen herkkiä mitokondriovirheille., Näiden solujen energiavajeen ja toksiinin kertymisen yhteisvaikutukset aiheuttavat todennäköisesti mitokondrioiden myopatioiden ja enkefalomyopatioiden tärkeimmät oireet.
yhteenvetona, riippuen ensisijainen geneettinen vika, mitokondrioiden sairaudet voivat aiheuttaa muutoksia seuraavat: hengitysteiden ketjun proteiineja, hengitysteiden ketjun avustavia proteiineja, mitokondrioiden RNA-käännös, mitokondrioiden sisäkalvon rasva miljöö, ehtyminen mitokondrio-DNA: ta, ja mitokondrioiden dynamiikkaa., Mitokondrioiden myopatiat voidaan jakaa myös luokkiin sen perusteella, mikä näistä prosesseista muuttuu.
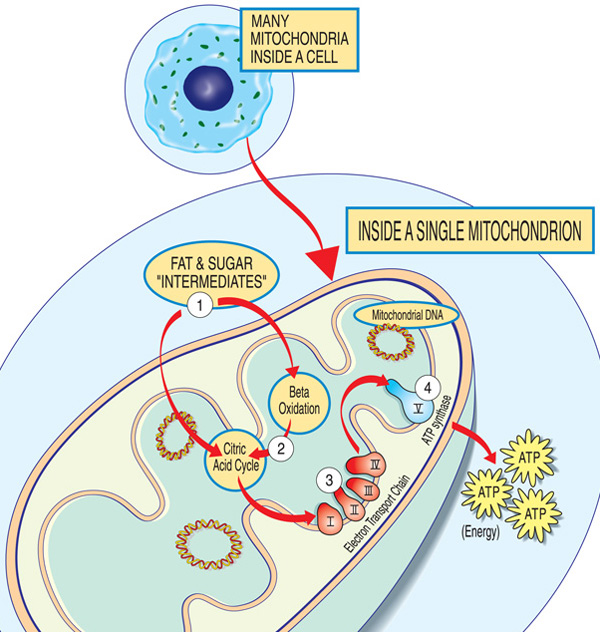
Jokainen mitochondrion on energiaa, että tehdas ”tuonti” sokerit ja rasvat, rikkoo ne alas ja ”vienti” energiaa (ATP: n) kautta seuraavasti: Rasvaa ja sokeria välituotteiden anna mitochondrion. Rasvahapot hajoavat beetaoksidaation ja elektronien poistumisen kautta sitruunahappokierrossa. Elektronit kulkevat elektroninkuljetusketjun suurimman kompleksin läpi. ATP: tä valmistaa ATP-syntaasi.,
mitkä ovat mitokondrioiden myopatioiden perintökuviot?
Mitokondrioiden geenit ovat monimutkaisia, ja usein mitokondrioiden tauti voi olla vaikea jäljittää läpi sukupuu. Mutta koska ne johtuvat viallisista geeneistä, mitokondriosairaudet kulkevat suvuissa.
ymmärtää, miten mitokondrioiden sairauksia siirretään läpi perheitä, se on tärkeää tietää, että on olemassa kahdenlaisia geenejä tärkeää mitokondrioita. Ensimmäinen tyyppi sijaitsee tumassa-solujemme osassa, joka sisältää suurimman osan geneettisestä materiaalistamme eli DNA: ta., Toinen tyyppi asuu yksinomaan mitokondrioiden sisällä olevassa DNA: ssa.
Mutaatiot joko tuman DNA: han (nDNA) tai mitokondrioiden DNA: ta (mtDNA) voi aiheuttaa mitokondrio-tauti.
useimmat nDNA (yhdessä mahdollisten mutaatioiden kanssa sillä on) periytyvät Mendeliläisessä kuviossa, mikä tarkoittaa löyhästi sitä, että yksi kopio kustakin geenistä tulee kummastakin vanhemmasta. Myös useimmat mitokondrioiden aiheuttamia sairauksia nDNA-mutaatioita (mukaan lukien Leigh oireyhtymä, MNGIE, ja jopa MDS) ovat autosomaalinen resessiivinen, mikä tarkoittaa, että se vie mutaatioita sekä kopioita geeni aiheuttaa sairauden.,
toisin Kuin nDNA, mtDNA kulkee vain äidiltä lapselle. Tämä johtuu siitä, että hedelmöityksen aikana, kun siittiöt sulautuvat munasoluun, sperman mitokondriot — ja sen mtDNA-tuhoutuvat. MtDNA-mutaatioiden aiheuttamat mitokondriosairaudet ovat siis ainutlaatuisia, koska ne periytyvät emomallilla (KS.kuva alla).
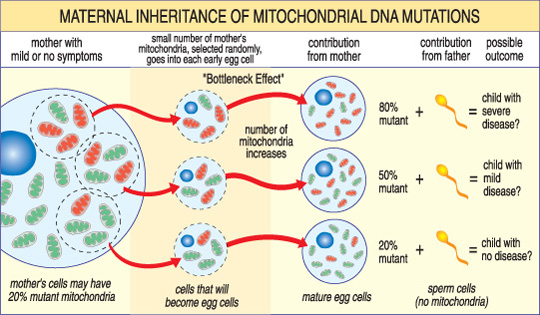
vakavuutta mitokondrioiden taudin lapsi riippuu prosenttiosuus epänormaali (mutantti) mitokondrioita munasolun, jotka muodostivat ne.,
Toinen ainutlaatuinen piirre mtDNA sairauksia johtuu siitä, että tyypillinen ihmisen solu — mukaan lukien muna solu — on vain yksi ydin, mutta satoja mitokondrioita. Yksittäinen solu voi sisältää sekä mutanttisia mitokondrioita että normaaleja mitokondrioita, ja niiden välinen tasapaino määrittää solun terveyden. Tämä auttaa selittämään, miksi mitokondriosairauden oireet voivat vaihdella niin paljon ihmisestä toiseen, jopa saman perheen sisällä.,
Kuvittele, että naisen munasolujen (ja muut solut hänen elin) sisältävät molemmat normaali ja mutantti mitokondrioita, ja että jotkut ovat vain muutaman mutantin mitokondrioita, kun taas toiset ovat monet. Lapsi tuli raskaaksi alkaen ”enimmäkseen terve” muna solu luultavasti ei kehittävät taudin, ja lapsi alkunsa alkaen ”enimmäkseen mutantti” muna solu luultavasti. Lisäksi naisella voi olla tai ei ole itse mitokondriosairauden oireita.
nämä sairaudet voivat myös syntyä satunnaisesti, eli niitä voi esiintyä ilman sukuhistoriaa.,
riski siirtää mitokondrioiden taudin lasten riippuu monista tekijöistä, mukaan lukien se, onko tauti on aiheuttanut mutaatioita nDNA tai mtDNA. Hyvä tapa saada lisätietoa näistä riskeistä on keskustella lääkärin tai geenineuvojan kanssa MDA: n hoitokeskuksessa. Katso myös faktoja genetiikasta ja Neuromuskulaarisista sairauksista.
Leave a Reply