årsager/arv
Hvad forårsager mitokondrielle sygdomme?
mitokondrielle myopatier er relativt almindelige. Primære mitokondrielle lidelser er de mest almindelige arvelige fejl i stofskiftet. Forekomsten af mitokondrielle encephalomyopatier til førskolebørn er 1 ud af 11.000. Mitokondriel sygdom forårsaget af mutationer i mitokondrielt DNA har en estimeret prævalens på 1 ud af 5.000. Mitokondriel sygdom forårsaget af mutationer i det nukleare DNA har imidlertid en estimeret prævalens på 1 ud af 35,000.,1
mitokondrielle sygdomme er ikke smitsomme, og de er ikke forårsaget af noget, en person gør. De er forårsaget af mutationer eller ændringer i gener — cellernes tegninger til fremstilling af proteiner.
gener er ansvarlige for at opbygge vores kroppe og overføres fra forældre til børn sammen med eventuelle mutationer eller defekter, de har. Det betyder, at mitokondrielle sygdomme er arvelige, selvom de ofte påvirker medlemmer af samme familie på forskellige måder. (For mere information om genetiske mutationer og mitokondriel sygdom, se nedenfor.,)
de gener, der er involveret i mitokondriesygdom, fremstiller normalt proteiner, der fungerer i eller på mitokondrierne. Inden for hver mitokondrie (ental af mitokondrierne), er disse proteiner udgør en del af et samlebånd, der bruger brændstof-molekyler, der stammer fra fødevarer til fremstilling af energi molekyle adenosin trifosfat (ATP) gennem en proces ved navn oxidative fosforylering. Denne meget effektive fremstillingsproces kræver ilt; uden for mitokondrionen er der mindre effektive måder at producere ATP uden ilt.,
proteiner i begyndelsen af mitokondriale samlebånd fungerer som lasthåndterere, der importerer brændstofmolekylerne — sukker og fedt — ind i mitokondrionen. Dernæst nedbryder andre proteiner sukker og fedtstoffer, ekstraherer energi i form af ladede partikler kaldet elektroner.
Proteiner mod slutningen af den linje — organiseret i fem grupper, som kaldes komplekser i, II, III, IV og V, og to mobile electron luftfartsselskaber, coenzym Q10 og cytochrom c — udnytte den energi fra disse elektroner til at lave ATP., Komplekser i gennem IV shuttle elektronerne ned linjen og kaldes derfor elektron transportkæden, og kompleks V faktisk junger ud ATP, så det kaldes også ATP syntase.
en mangel i et eller flere af disse komplekser er den typiske årsag til en mitokondriel sygdom. (Faktisk er mitokondrielle sygdomme undertiden opkaldt efter en specifik mangel, såsom kompleks i-mangel.) Coenzym Q10-mangel på grund af nukleare DNA-mutationer kan præsentere med proksimal muskelsvaghed i isolation., For at fungere korrekt skal proteinerne fra den o .idative phosphoryleringsvej oversættes, importeres til mitokondrierne og indsættes i den indre mitokondrielle membran. Mutationer i gener, der påvirker disse processer, kan også forårsage mitokondriel myopati.
defekter i gener, der er relateret til mitokondriernes struktur og dynamik, kan også være involveret i udviklingen af myopatier., Når en celle er fyldt med defekte mitokondrier, bliver den ikke kun berøvet ATP, den kan også akkumulere en efterslæb af ubrugte brændstofmolekyler og ilt med potentielt katastrofale virkninger.
i sådanne tilfælde bruges overskydende brændstofmolekyler til at fremstille ATP på ineffektive måder, hvilket kan generere potentielt skadelige biprodukter, såsom mælkesyre. (Dette sker også, når en celle har en utilstrækkelig iltforsyning, hvilket kan ske med muskelceller under anstrengende træning.,) Ophobningen af mælkesyre i blodet – kaldet mælkesyreacidose-er forbundet med muskeltræthed og kan faktisk skade muskler og nervevæv.
i mellemtiden kan ubrugt ilt i cellen omdannes til destruktive forbindelser kaldet reaktive o .ygenarter, herunder såkaldte frie radikaler. (Dette er målene for antio .idant medicin og vitaminer.)
ATP afledt af mitokondrier giver den vigtigste kilde til magt til muskelcellekontraktion og nervecellefyring. Så muskelceller og nerveceller er særligt følsomme over for mitokondrielle defekter., De kombinerede virkninger af energiberøvelse og toksinakkumulering i disse celler giver sandsynligvis anledning til de vigtigste symptomer på mitokondrielle myopatier og encephalomyopatier.
I resumé, afhængigt af den primære genetiske defekt, mitokondrielle sygdomme, der kan være forårsaget af ændringer af følgende: respiratorisk kæde proteiner, respiratorisk kæde accessoriske proteiner, mitokondrielle RNA oversættelse, mitochondrial indre membran lipid miljø, udtømning af mitokondrie-DNA, og mitokondrie-dynamik., Mitokondrielle myopatier kan også opdeles i kategorier baseret på hvilke af disse processer der ændres.
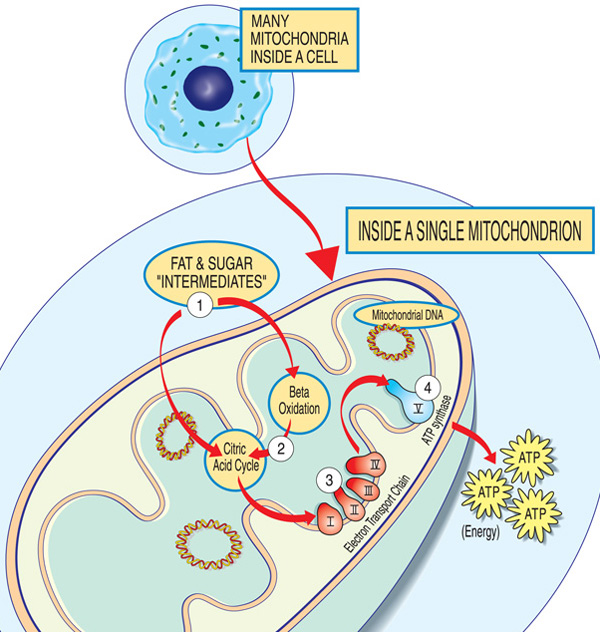
Hver mitokondrie er en energi-fabrik, der “import” sukker og fedt, bryder dem ned, og “eksport” energi (ATP) ved hjælp af følgende fremgangsmåde: sukker og Fedt mellemprodukter indtaste mitokondrier. Fedtsyrer nedbrydes gennem beta-o .idation og fjernelse af elektroner i citronsyrecyklussen. Elektroner passeres gennem det store kompleks af elektrontransportkæden. ATP er lavet af ATP syntase.,
Hvad er arvemønstrene i mitokondrielle myopatier?
mitokondriel genetik er kompleks, og ofte kan en mitokondriel sygdom være vanskelig at spore gennem et stamtræ. Men fordi de er forårsaget af defekte gener, kører mitokondrielle sygdomme i familier.
for at forstå, hvordan mitokondrielle sygdomme overføres gennem familier, er det vigtigt at vide, at der er to typer gener, der er essentielle for mitokondrier. Den første type er indeholdt i kernen — den del af vores celler, der indeholder det meste af vores genetiske materiale eller DNA., Den anden type ligger udelukkende inden for DNA indeholdt i selve mitokondrierne.
mutationer i enten nuklear DNA (nDNA) eller mitokondrielt DNA (mtDNA) kan forårsage mitokondriel sygdom.
de fleste nDNA (sammen med eventuelle mutationer det har) er nedarvet i en Mendelian mønster, løst betyder, at en kopi af hvert gen kommer fra hver forælder. De fleste mitokondrielle sygdomme forårsaget af nDNA-mutationer (inklusive Leigh syndrom, mngie og endda MDS) er autosomale recessive, hvilket betyder, at det tager mutationer i begge kopier af et gen for at forårsage sygdom.,
i modsætning til nDNA passerer mtDNA kun fra mor til barn. Det skyldes, at sædets mitokondrier — og dets mtDNA — ødelægges under undfangelsen, når sædcellen smelter sammen med ægget. Mitokondrielle sygdomme forårsaget af mtDNA-mutationer er således unikke, fordi de er arvet i et moderligt mønster (se illustration nedenfor).
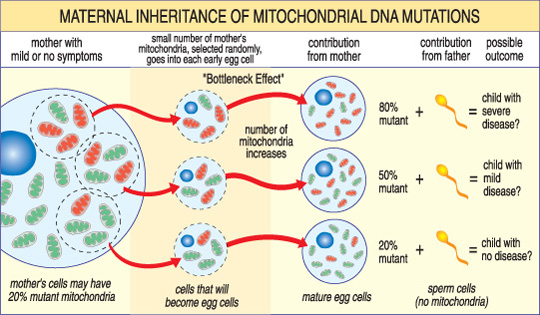
sværhedsgraden af en mitokondriel sygdom hos et barn afhænger af procentdelen af unormale (mutante) mitokondrier i ægcellen, der dannede dem.,
et andet unikt træk ved mtDNA — sygdomme stammer fra det faktum, at en typisk human celle — inklusive ægcellen-kun indeholder en kerne, men hundreder af mitokondrier. En enkelt celle kan indeholde både mutante mitokondrier og normale mitokondrier, og balancen mellem de to vil bestemme cellens helbred. Dette hjælper med at forklare, hvorfor symptomerne på mitokondriel sygdom kan variere så meget fra person til person, selv inden for samme familie.,
Forestil dig, at en kvindes ægceller (og andre celler i hendes krop) indeholder både normale og mutante mitokondrier, og at nogle kun har nogle få mutante mitokondrier, mens andre har mange. Et barn undfanget fra en “for det meste sund” ægcelle ville sandsynligvis ikke udvikle sygdom, og et barn undfanget fra en “for det meste mutant” ægcelle vil sandsynligvis. Også kvinden kan eller måske ikke have symptomer på mitokondriel sygdom selv.
disse sygdomme kan også opstå sporadisk, hvilket betyder, at de kan forekomme uden familiehistorie.,
risikoen for at overføre en mitokondriel sygdom til børn afhænger af mange faktorer, herunder om sygdommen er forårsaget af mutationer i nDNA eller mtDNA. En god måde at finde ud af mere om disse risici er at tale med en læge eller genetisk rådgiver på et MDA-Plejecenter. Se også fakta om genetik og neuromuskulære sygdomme.
Leave a Reply